|
 
 |
|
BRIEF REPORT |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 1 | Page : 78-83 |
| |
Polymerase chain reaction optimization for amplification of Guanine-Cytosine rich templates using buccal cell DNA
C. H. W. M. R. Chandrasekara Bhagya1, WS Wijesundera Sulochana1, N Perera Hemamali2
1 Department of Biochemistry and Molecular Biology, Faculty of Medicine, University of Colombo, Sri Lanka
2 Department of Psychological Medicine, Faculty of Medicine, University of Colombo, Sri Lanka
| Date of Web Publication | 4-Jun-2013 |
Correspondence Address:
C. H. W. M. R. Chandrasekara Bhagya
Department of Biochemistry and Molecular Biology, Faculty of Medicine, University of Colombo, P. O. Box 271, Kynsey Road, Colombo
Sri Lanka
 Source of Support: National Science Foundation, Conflict of Interest: None
DOI: 10.4103/0971-6866.112898

 Abstract Abstract | | |
Context: Amplification of Guanine-Cytosine (GC) -rich sequences becomes important in screening and diagnosis of certain genetic diseases such as diseases arising due to expansion of GC-rich trinucleotide repeat regions. However, GC-rich sequences in the genome are refractory to standard polymerase chain reaction (PCR) amplification and require a special reaction conditions and/or modified PCR cycle parameters.
Aim: Optimize a cost effective PCR assay to amplify the GC-rich DNA templates.
Settings and Design: Fragile X mental retardation gene (FMR 1) is an ideal candidate for PCR optimization as its GC content is more than 80%. Primers designed to amplify the GC rich 5' untranslated region of the FMR 1 gene, was selected for the optimization of amplification using DNA extracted from buccal mucosal cells.
Materials and Methods: A simple and rapid protocol was used to extract DNA from buccal cells. PCR optimization was carried out using three methods, (a) substituting a substrate analog 7-deaza-dGTP to dGTP (b) in the presence of a single PCR additive and (c) using a combination of PCR additives. All PCR amplifications were carried out using a low-cost thermostable polymerase.
Results: Optimum PCR conditions were achieved when a combination of 1M betaine and 5% dimethyl sulfoxide (DMSO) was used.
Conclusions: It was possible to amplify the GC rich region of FMR 1 gene with reproducibility in the presence of betaine and DMSO as additives without the use of commercially available kits for DNA extraction and the expensive thermostable polymerases.
Keywords: Enhancers, fragile X syndrome, guanine-cytosine-rich sequences, polymerase chain reaction additive, polymerase chain reaction
How to cite this article:
Bhagya CC, Wijesundera Sulochana W S, Hemamali N P. Polymerase chain reaction optimization for amplification of Guanine-Cytosine rich templates using buccal cell DNA. Indian J Hum Genet 2013;19:78-83 |
How to cite this URL:
Bhagya CC, Wijesundera Sulochana W S, Hemamali N P. Polymerase chain reaction optimization for amplification of Guanine-Cytosine rich templates using buccal cell DNA. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:78-83. Available from: http://www.ijhg.com/text.asp?2013/19/1/78/112898 |
| Introduction | |  |
Polymerase chain reaction (PCR) is a widely used technique in many laboratories for diagnostic purposes and molecular biology studies. Most of the DNA templates do not require special conditions to undergo amplification, when the deoxyribonucleotide content is equally distributed among the length of the fragment to be amplified. However, lack of amplification efficiency and non-specific amplification may result in the presence of impurities and inhibitors. Furthermore, PCR amplification of Guanine-Cytosine (GC) rich regions of genomic DNA is difficult. GC rich regions produce complex inter and intra strand folding (hairpins and loops) due to increased hydrogen bonding with neighboring cytosine and guanine. [1] These secondary structures in DNA are resistant to melting and cause Taq DNA polymerases to stall and also hampers primer annealing, resulting in incomplete or non-specific amplification. [2],[3]
Different methods have been developed to improve the amplification of GC rich sequences. These include addition of organic substances (additives), use of modified dNTPs and modification of thermal cycling program in PCR. [2] The additives improve the amplification by unwinding the double stranded DNA (dsDNA) helix and thereby reducing the melting temperature. The most prominent PCR enhancing additives currently used are either betaine, small sulfoxides such as dimethyl sulfoxide (DMSO), formamide or reducing compounds such as beta-mercaptoethanol or dithiothreitol. [1],[4],[5],[6] Addition of DMSO, formamide or glycerol denatures dsDNA. Betaine, an amino acid analog, stabilizes the denatured DNA. [7],[8] Substituting, the guanine base analog, 7-deaza-dGTP for dGTP, in a ratio of 3:1 can reduce the number of hydrogen bonds that are formed between guanine and cytosine in dsDNA as well as single stranded DNA (ssDNA) because it lacks nitrogen at the seventh position of the purine ring. Therefore, addition of 7-deaza-dGTP prevents formation of stable intramolecular G * C Hoogsteen base pairing without disrupting the normal Watson-Crick base pairing. [1],[9],[10] Alkaline denaturation of template prior to PCR is also employed to facilitate the PCR. [11]
The above facts are relevant in genetic studies as 3% of the human genome is highly GC rich and 28% of the genes are located within the GC rich regions. [11] Several genetic diseases arise due to the expansion of GC rich trinucleotide repeat sequences. One such example is the fragile X mental retardation gene (FMR 1), which contains a polymorphic CGG repeat of 5-55 copies in the 5' region of the gene. Fragile X Syndrome occurs due to expansion of these CGG trinucleotide repeats up to more than 200 copies. [4] The Huntingtin gene associated with Huntington disease contains a polymorphic CAG trinuleotide repeats with varying the repeat number from 11 to 34 CAG. [12] Myotonic dystrophy protein kinase gene is another GC rich gene which contains CTG repeats and involve in the myotonic dystrophy. [13] Genetic tests for screening and disease diagnosis associated with expansion of trinucleotide repeats are based on identifying these repeat sequences.
PCR amplification has been proposed as a rapid method for amplification of trinucleotide repeat regions compared to Southern hybridization. In this article, we report the results of three methods that were used to optimize the PCR amplification of the GC rich 5' region of the FMR 1 gene using DNA extracted from buccal cells. The aim was to develop a low-cost PCR assay for amplification of GC rich regions which can be used to screen diseases associated with GC rich sequences.
| Materials and Methods | | |
All chemicals used in the study were molecular grade and unless otherwise specified, were purchased from Sigma Chemical Company (St Louis, MO, USA). Taq DNA polymerase, 5 × buffer, MgCl 2 and dNTP were purchased from Promega Company (Madison, WI, USA). Primers (Oligonucleotides) were custom synthesized from Vivantis (Singapore). Amplification was performed in a Bio Rad/My cycler thermal cycler.
Ethical approval for the study was granted by the Ethics Review Committee of the University of Colombo, Faculty of Medicine.
Method described by Handel et al. (2006) with minor modifications was used to extract DNA from buccal cells. Persons with diagnosed genetic disorders were excluded. Informed and consenting subjects (age 5-12 years) were refrained from eating and drinking for 30 min prior to sample collection. The mouths of the subjects were rinsed and buccal cells were collected by rolling an autoclaved cotton bud inside the cheek twenty times on each side. Cotton bud containing the buccal cells was dipped in a microcentrifuge tube containing 500 μl of lysis buffer which has a composition of 0.1 × Gitschiez buffer (10 × GB containing 670 mmTris pH 8.0, 166 mm (NH 4 ) 2 SO 4 and 67 mm MgCl 2 ) and 0.5% Triton × 100. [14] The collected buccal cells were resuspended in lysis buffer by rotating the cotton bud clockwise and anti-clockwise five times in each direction and then pressed against the wall of the microcentrifuge tube. The buccal cells resuspended in lysis buffer was stored at −20°C until DNA extraction.
For extraction, the stored buccal cell sample was allowed to reach room temperature before vortexing for 10 s. Proteinase K was added to reach final concentration of 40 μg/ml, mixed well and incubated at 63°C for 6 min. Thereafter, 250 μl of 4.5 M NaCl was added, mixed vigorously, centrifuged at 13000 g for 15 min and the supernatant was recovered. The genomic DNA in the supernatant was ethanol precipitated and resuspended in 20 μl of of Tris- EDTA pH 8.0. The concentration and the integrity of the extracted DNA was determined by ethidium bromide stained 0.8% agarose gel electrophoresis and compared against known concentrations of lambda DNA.
CGG repeats spanning around 30 repeats (~300 bp) of the FMR1 gene 5' region was selected for PCR amplification using primer c and f as stated in Fu et al. (1991). [9] The following three procedures (a), (b) and (c) were carried out to amplify the GC rich template. DNA extracted from a single individual was used in all optimization reactions and hot start PCR was performed. Ten microliters from all PCR products were electrophoresed on 2% agarose gel containing 10 μg/ml of ethidium bromide for 30 min at 75 V and bands were visualized under ultraviolet light from Uvi pro silver gel documentation system (UV tech).
Using substrate analogue, 7-deaza-dGTP
- Initially the PCR was carried out as described by Fu et al. (1991) by partially substituting 7-deaza-dGTP for dGTP at a ratio of 3:1. The 15 μl reaction mixture contained l × PCR buffer, 2 mM MgCl 2 , 150 μM deaza dGTP, 200 μM dATP, 200 μm dCTP, 200 μM dTTP, 50 μM dGTP, 10% DMSO, 2 μM of each primer and 0.75 U Taq DNA polymerase. Thermo cycle condition was: Initial denaturation at 95°C for 10 min followed by 25 cycles of denaturation at 95°C for 1.5 min, annealing at 65°C for 1 min and extension at 72°C for 2 min. [9]
- The annealing temperature was varied from 58°C to 65°C by a 1°C increment keeping the reaction conditions the same as in (1) above.
- The reaction conditions were varied as follows, template DNA (10-150 ng/μl by 10 ng/μl increments), MgCl 2 (1-4 mM by 0.5 increments), dNTP (100 μm-500 μM by 100 μm increments, primer (0.1-1 μm by 0.2 μM increments), and Taq DNA polymerase (0.5 U-2 U by 0.5 U increments). The 7-deaza-dGTP and the DMSO concentration were kept at 150 μm and 10% respectively. The thermal cycling conditions were as in (1) above.
Using PCR additivesPCR optimizations were performed in the presence of several PCR additives separately. The concentrations of the additives were varied as shown in [Table 1]. The 25 μl reaction mixture contained l × PCR buffer, 2 mM MgCl 2 , and 200 μM dNTP, 2 μM each primer and 0.75 U Taq DNA polymerase and thermo cycling parameters were as in (a) 1.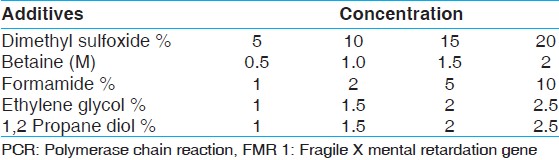 | Table 1: Different PCR additives and their concentrations used for PCR amplification of FMR 1 gene
Click here to view |
Using combination of additives
Taking into consideration the results obtained from (b) above, different combinations of betaine and DMSO were tested in a 25 μl volume containing, 0.2 mM dNTPs, 1.5 mM MgCl 2, 1 × PCR buffer, 1 U Taq DNA polymerase, 0.1 μM of each primer and 50 ng of genomic DNA. Thermo cycle condition was: Initial denaturation at 95°C for 10 min followed by 25 cycles of denaturation at 95°C for 1.5 min, annealing at 65°C for 1 min and extension at 72°C for 2 min.
- The concentration of betaine was kept constant at 1.0 M and varied the concentration of DMSO (5%, 10%, 15% and 20%).
- The concentration of DMSO (5%) was kept constant and varied the concentration of betaine (0.5 M, 1 M, 1.5 M, 2 M and 2.2 M).
- With the optimised betaine (1 M) and DMSO, (5%) concentrations, the PCR amplification was performed using thermal cycle parameters sated by Baskeran et al. (1996): Denaturation at 94°C for 15 s, annealing at 65°C for 2 min, and extension at 68°C for 5 min for 40 cycles. A series of DNA concentrations were tested using these conditions (10 ng, 15 ng, 25 ng). [15]
- Further optimization of the cycle parameters was carried out by decreasing the annealing time from 2 min to 1 min and the extension time from 5 min to 2 min. The PCR cycles were also reduced from 40 cycles to 30 cycles.
After optimizing the PCR conditions and thermal cycle parameters, PCR was performed for the remaining samples using DNA extracted from buccal cells.
| Results | | |
All attempts to amplify the GC rich region of the FMR 1 gene fragment using protocol (a) by partially substituting 7-deaza-dGTP for dGTP in a ratio of 3:1, varying the reaction conditions and the annealing temperature failed and no amplification was observed (data not presented).
In protocol (b) PCR amplification was carried out in the presence of different concentrations of various additives separately. Addition of 1, 2 propane diol and ethylene glycol did not promote amplification at any given concentration (data not presented). However, non-specific amplifications were observed in the presence of 5% formamide, 5% DMSO and 1 M betaine (data not shown).
As a single PCR additive was unable to produce the desired product, combination of two additives were tested. Non-specific amplification was reduced in the presence of 1M betaine and 5% DMSO when the PCR was carried out according to the method described in protocol (c) 1 and 2 (data not presented). Therefore, further optimization was performed in the presence of 1 M betaine and 5% DMSO Protocol (c) 3 and the desired product (around 300 bp) was obtained with a non-specific amplification [Figure 1].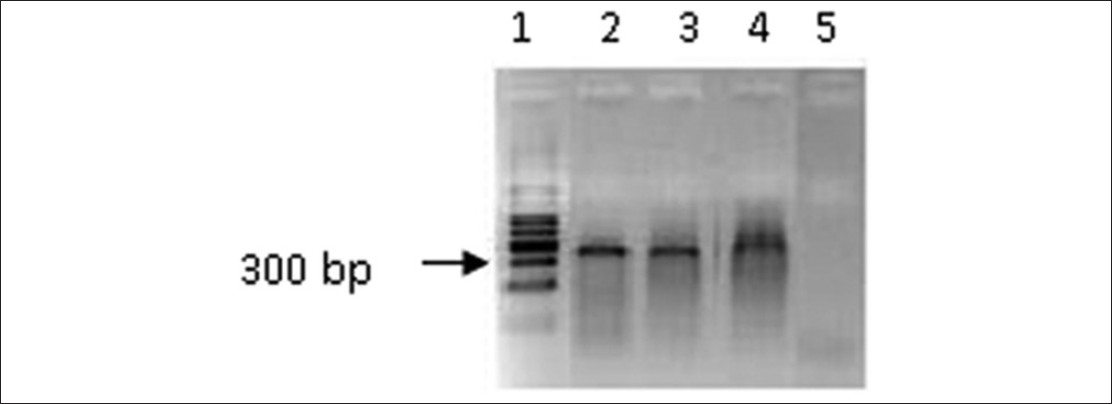 | Figure 1: Polymerase chain reaction amplification of Fragile X mental retardation gene 5' region using a serial dilution of genomic DNA in the presence of betaine (1.0 M) and dimethyl sulfoxide (5%). Lane 1: 100 bp ladder, Lane 2: 10 ng, Lane 3: 15 ng, Lane 4: 25 ng, Lane 5: Negative control (without DNA). The cycle conditions: Initial denaturation 94°C for 15 s followed by 40 cycles of denaturation at 94°C for 15 s, annealing at 65°C for 2 min, and extension at 68°C for 5 min for 40 cycles
Click here to view |
In order to improve the results annealing time, extension time and number of cycles of the PCR was reduced. The optimum assay conditions were: 0.2 mM dNTPs, 1.5 mM MgCl 2, 1 × PCR buffer, 1 U Taq DNA polymerase, 0.1 μM of each primer, 50 ng of genomic DNA, betaine (1M) and DMSO (5%) in a 25 μl reaction volume with an initial denaturation at 94°C for 15 s followed by 30 cycles of denaturation at 94°C for 15 s annealing at 65°C for 1 min, and extension at 68°C for 2 min.
[Figure 2] shows PCR amplification carried out under optimum assay conditions on representative samples of buccal cell DNA. It was observed that buccal cell DNA extracts (prior to ethanol precipitation) could also be used under optimum conditions to amplify the GC rich region with reproducibility.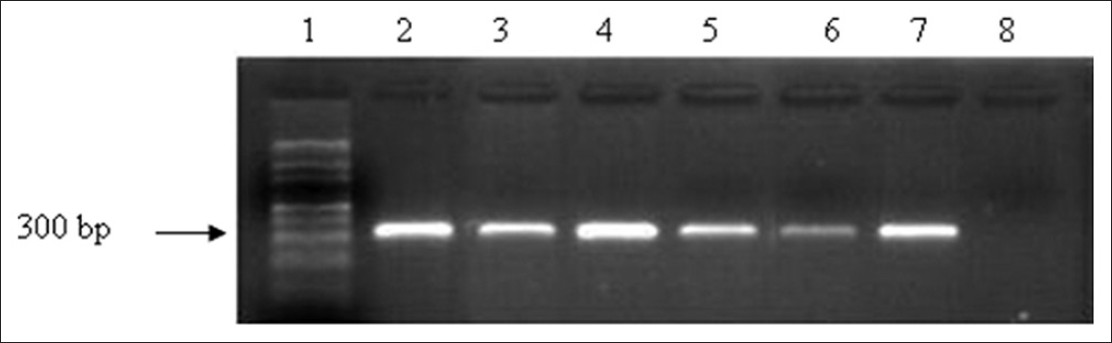 | Figure 2: Polymerase chain reaction amplification of Fragile X mental retardation gene 5' region under optimized conditions using buccal cell DNA. Lane 1:100 bp ladder, Lane 2 - 7: Representative samples (50 ng), Lane 8: Negative control (without DNA)
Click here to view |
| Discussion | | |
PCR amplification of the 5' untranslated CGG repeat region of the FMR I gene is used as a rapid and reliable method for screening of fragile X syndrome. The GC content of the gene is very high (>80%), and standard PCR cannot be performed. Therefore, we selected the FMR I gene fragment for PCR optimization. Concentration dependent combination of different PCR additives with modified thermocycle conditions were tested for a reliable and low cost PCR assay.
Amplification of GC rich templates including the FMR1 gene have been reported by many scientists using expensive thermostable polymerases with additives [9],[16],[17],[18],[19],[20] or with a combination of thermostable polymerases with additives [15],[21],[22] or using expensive commercially available kits. [23],[24] Commercially available PCR enhancer solutions such as Q solution (Qiagen), [5] PCR enhancer solution (Invitrogen), [20] GC-melt™ reagent (BD Biosciences Clontech, Palo Alto, CA) [3] and Hi-Spec PCR additive (Bioline) [5] have also been used to promote amplification of GC rich templates, which are too costly for laboratories in low income countries. Additionally, commercially available kits have been used in DNA extractions procedures, further increasing the cost per PCR assay.
In the initial procedure, the use of 7-deaza-dGTP even with altering the PCR conditions and cycle parameters did not allow successful amplification. A possible reason could be the type of thermostable polymerase that was used in the present study. Similar results where complete absence of PCR products were also observed by Saluto et al. (2005) in their study of amplification of GC rich templates using 7-deaza-dGTP.
PCR amplification using a combination of DMSO (5%) and betaine (1 M) worked effectively as DMSO facilitate unwinding of the dsDNA and resulting ssDNA are stabilized by betaine. DMSO and betaine combination have been used in several protocols in the amplification of GC rich templates under different concentration of the additives and/or PCR thermocycle conditions and different thermostable DNA polymerases. [9],[10],[21],[22],[25] Baskeran et al. (1996) reported the amplification of FMR1 gene fragment using the same primers (c and f) but with a slightly higher concentration betaine, (1.1 M) and DMSO (7%) or 10% DMSO or 2.2 M betiane or 5% DMSO and 2.0 M betaine. We found that betaine concentration higher than 1.5 M inhibited the PCR. Furthermore, addition of betaine and DMSO does not interfere with PCR product visualization using ethidium bromide stained agarose gels unlike when 7-deaza-dGTP is used in amplification where alternate detections methods have to be used. [10],[15]
In the present study, buccal cells were collected for DNA extraction as a non-invasive method. It is an excellent source of DNA for diagnosis and large-scale molecular epidemiological studies. The DNA extraction procedure was simple, rapid (1½ h) and cost effective when compared to DNA extraction from blood and the use of commercially available DNA extraction kits. Both precipitated DNA and DNA extract prior to DNA precipitation was suitable for PCR as it was observed that both templates gave bands with equal intensity after amplification, confirming the suitability of the DNA extraction method. It was also observed that the DNA extract stored at −20°C for more than 6 months could be used for amplification under optimized PCR conditions.
We were successful in optimizing the PCR without the use of commercially available expensive kits and thermostable polymerases to amplify GC rich templates. It was possible to obtain reproducible results with a low cost thermostable polymerase in the presence of betaine and DMSO as additives and the amplified products could be easily visualized by ethidium bromide stained agarose gel electrophoresis. The application of this optimized protocol can thus be recommended as a low cost, and reliable means to amplify GC rich DNA templates in laboratories in low resource settings.
| Acknowledgments | | |
This work has been funded by the National Science Foundation Sri Lanka. (Grant No: RG/2009/HS/02).
| References | | |
| 1. | Musso M, Bocciardi R, Parodi S, Ravazzolo R, Ceccherini I. Betaine, dimethyl sulfoxide, and 7-deaza-dGTP, a powerful mixture for amplification of GC-rich DNA sequences. J Mol Diagn 2006;8:544-50. 
|
| 2. | Shore R, Paul N. Robust PCR amplification of GC-rich targets with hot start 7-deaza-dGTP. TriLink BioTechnol 2010;49:841-3.
|
| 3. | Hubé F, Reverdiau P, Iochmann S, Gruel Y. Improved PCR method for amplification of GC-rich DNA sequences. Mol Biotechnol 2005;31:81-4.
|
| 4. | Bachmann HS, Siffert W, Frey UH. Successful amplification of extremely GC-rich promoter regions using a novel 'slowdown PCR' technique. Pharmacogenetics 2003;13:759-66.
|
| 5. | Ralser M, Querfurth R, Warnatz HJ, Lehrach H, Yaspo ML, Krobitsch S. An efficient and economic enhancer mix for PCR. Biochem Biophys Res Commun 2006;347:747-51.
|
| 6. | Zhang Z, Yang X, Meng L, Liu F, Shen C, Yang W. Enhanced amplification of GC-rich DNA with two organic reagents. Biotechniques 2009;47:775-9.
|
| 7. | Jensen MA, Fukushima M, Davis RW. DMSO and betaine greatly improve amplification of GC-rich constructs in de novo synthesis. PLoS One 2010;5:e11024.
|
| 8. | Henke W, Herdel K, Jung K, Schnorr D, Loening SA. Betaine improves the PCR amplification of GC-rich DNA sequences. Nucleic Acids Res 1997;25:3957-8.
|
| 9. | Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991;67:1047-58.
|
| 10. | Jung A, Ruckert S, Frank P, Brabletz T, Kirchner T. 7-Deaza-2'- deoxyguanosine allows PCR and sequencing reactions from CpG islands. Mol Pathol 2002;55:55-7.
|
| 11. | Mamedov TG, Pienaar E, Whitney SE, TerMaat JR, Carvill G, Goliath R, et al. A fundamental study of the PCR amplification of GC-rich DNA templates. Comput Biol Chem 2008;32:452-7.
|
| 12. | Rosales-Reynoso MA, Vilatela EA, Ojeda RM, Arce-Rivas A, Sandoval L, Troyo-Sanromán R, et al. PCR approach for detection of fragile X syndrome and huntington disease based on modified DNA: Limits and utility. Genet Test 2007;11:153-9.
|
| 13. | López Castel A, Nakamori M, Tomé S, Chitayat D, Gourdon G, Thornton CA, et al. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum Mol Genet 2011;20:1-15.
|
| 14. | Handel CM, Pajot LM, Talbot SL, Sage GK. Use of buccal swabs for sampling DNA from nestling and adult birds. Wildl Soc Bull 2006;34:150-5.
|
| 15. | Baskaran N, Kandpal RP, Bhargava AK, Glynn MW, Bale A, Weissman SM. Uniform amplification of a mixture of deoxyribonucleic acids with varying GC content. Genome Res 1996;6:633-8.
|
| 16. | Florencia G, Irene S, Veronica F. Fragile-X mental retardation: Molecular diagnosis in Argentine patients. J Biochem Mol Biol 2006;39:766-73.
|
| 17. | Saluto A, Brussino A, Tassone F, Arduino C, Cagnoli C, Pappi P, et al. An enhanced polymerase chain reaction assay to detect pre-and full mutation alleles of the fragile X mental retardation 1 gene. J Mol Diagn 2005;7:605-12.
|
| 18. | Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn 2008;10:43-9.
|
| 19. | Khaniani MS, Kalitsis P, Burgess T, Slater HR. An improved diagnostic PCR assay for identification of cryptic heterozygosity for CGG triplet repeat alleles in the fragile X gene (FMR1). Mol Cytogenet 2008;1:5.
|
| 20. | O'Connell CD, Atha DH, Jakupciak JP, Amos JA, Richie Kl. Standardization of PCR amplification for fragile X trinucleotide repeat measurements. Clin Genet 2002;61:13-20.
|
| 21. | Hamdan H, Tynan JA, Fenwick RA, Leon JA. Automated Detection of Trinucleotide Repeats in Fragile X Syndrome. Mol Diagn 1997;2:259-69.
|
| 22. | Chen L, Hadd A, Sah S, Filipovic-Sadic S, Krosting J, Sekinger E, et al. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J Mol Diagn 2010;12:589-600.
|
| 23. | Dutta S, Das M, Bhowmik AD, Sinha S, Chattopadhyay A, Mukhopadhyay K. Screening of rural children in West Bengal for fragile-X syndrome. Indian J Med Res 2009;130:714-9.
[PUBMED]  |
| 24. | Fernandez-Carvajal I, Walichiewicz P, Xiaosen X, Pan R, Hagerman PJ, Tassone F. Screening for expanded alleles of the FMR1 gene in blood spots from newborn males in a Spanish population. J Mol Diagn 2009;11:324-9.
|
| 25. | Frackman S, Kobs G, Simpson D, Storts D. Betaine and DMSO: Enhancing agents for PCR. Promega Notes 1998;65:27.
|
[Figure 1], [Figure 2]
[Table 1]
|










