|
 
 |
|
ORIGINAL ARTICLE |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 2 | Page : 251-258 |
| |
Comparison of in-vitro and in-vivo response to fetal hemoglobin production and γ-mRNA expression by hydroxyurea in Hemoglobinopathies
Khushnooma Italia1, Farah Jijina2, Rashid Merchant3, Suchitra Swaminathan1, Anita Nadkarni1, Maya Gupta1, Kanjaksha Ghosh1, Roshan Colah1
1 National Institute of Immunohematology, Hematogenetics, 13th Floor, K.E.M. Hospital Campus, Parel, Mumbai, India
2 Department of Hematology, 10th Floor, K.E.M. Hospital, Parel, Mumbai, India
3 Department of Pediatrics, Nanavati Hospital, Vileparle (W), Mumbai, India
| Date of Web Publication | 5-Aug-2013 |
Correspondence Address:
Roshan Colah
National Institute of Immunohematology, 13th Floor, N. M. S. Bldg, K.E.M. Hospital Campus, Parel, Mumbai - 400 012
India
 Source of Support: None, Conflict of Interest: None
PMID: 24019630 
 Abstract Abstract | | |
Background: Hydroxyurea, which induces Fetal hemoglobin (HbF) synthesis, is the only drug widely used in different hemoglobinopathies; however, the response is very variable. We compared the efficacy of hydroxyurea in-vitro in erythroid cultures and in-vivo in the same patients with different hemoglobinopathies to induce HbF production and enhance γ-messenger RNA expression.
Materials and Methods: A total of 24-patients with different Hemoglobinopathies were given hydroxyurea and their response was studied in-vivo and in-vitro on mononuclear cells collected from them simultaneously.
Results: A total of 57.7% of patients (responders) showed no further crisis or transfusion requirements after hydroxyurea therapy with a mean increase in fetal cells (F-cells) of 63.8 ± 59.1% and γ-mRNA expression of 205.5 ± 120.8%. In-vitro results also showed a mean increase in F-cells of 27.2 ± 24.7% and γ-mRNA expression of 119.6% ± 65.4% among the treated cells. Nearly 19.0% of the partial-responders reduced their transfusion requirements by 50% with a mean increase in F-cells of 61.2 ± 25.0% and 28.4 ± 25.3% and γ-mRNA-expression of 21.0% ± 1.4% and 80.0% ± 14.1% in-vivo and in-vitro respectively. The non-responders (15.3%) showed no change in their clinical status and there was no significant increase in F-cells levels and γ-mRNA expression in-vivo or in-vitro.
Conclusion: Thus, this method may help to predict the in-vivo response to hydroxyurea therapy; however, a much larger study is required.
Keywords: 2 phase liquid erythroid cell culture, hydroxyurea, sickle cell disease, β-thalassemia syndromes, γ-mRNA expression
How to cite this article:
Italia K, Jijina F, Merchant R, Swaminathan S, Nadkarni A, Gupta M, Ghosh K, Colah R. Comparison of in-vitro and in-vivo response to fetal hemoglobin production and γ-mRNA expression by hydroxyurea in Hemoglobinopathies. Indian J Hum Genet 2013;19:251-8 |
How to cite this URL:
Italia K, Jijina F, Merchant R, Swaminathan S, Nadkarni A, Gupta M, Ghosh K, Colah R. Comparison of in-vitro and in-vivo response to fetal hemoglobin production and γ-mRNA expression by hydroxyurea in Hemoglobinopathies. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:251-8. Available from: http://www.ijhg.com/text.asp?2013/19/2/251/116128 |
| Introduction | |  |
Inherited disorders of hemoglobin, such as the structural Hemoglobinopathies, and the thalassemia syndromes are common, affecting around 7% of the global population with significant clinical conditions. [1] In India, the prevalence of sickle cell carriers ranges from 2% to 34% in different population groups [2],[3] whereas that of β-thalassemia ranges from 1% to 17%. [4],[5] On the other hand, Hemoglobin E (HbE) is generally found in the eastern and north eastern region of the country with a frequency of 3-10% and 4-51% respectively. [6]
Although, sickle cell disease has a milder clinical presentation than in Africans among tribal groups in India, many patients particularly from the non-tribal groups suffer from severe forms of the disease leading to repeated vaso-occlusive crisis, severe anemia, and end organ damage. [7],[8] So far, mainly supportive measures have been used for decreasing the severity of symptoms. Few children with the severe forms of β-thalassemia receive regular blood transfusions and adequate iron chelation, the mainstay of treatment, due to the high-cost involved for optimum management. [5] Thus, there was a need for an efficacious drug with low toxicity, which could reduce the clinical symptoms of these Hemoglobinopathies in a cost-effective way.
Hydroxyurea reduces the frequency of vaso-occlusive crisis and blood transfusion requirements in sickle cell anemia patients [8],[9] as well as the transfusion requirements in some β-thalassemia patients. [10],[11] As there are no clear clinical or genetic factors, which can predict the response of these patients to hydroxyurea therapy, patients who are non-responders also get exposed to the drug. Thus, this study thus investigated whether the response to hydroxyurea in erythroid cultures of patients with different Hemoglobinopathies to increase fetal hemoglobin (HbF) and γ-gene expression in-vitro correlates simultaneously with the in-vivo response to hydroxyurea among the same patients.
| Materials and Methods | | |
Out of 24 patients (8 males and 16 females; age 5-30 years) with different Hemoglobinopathies (severe sickle cell anemia patients - 6; severe sickle-β-thalassemia patients - 2, these patients had more than 5 episodes of painful vaso-occlusive crisis and occasional blood transfusion requirements; β-thalassemia intermedia patients - 5, who presented after 2 years of age and required occasional blood transfusions initially however, later they became transfusion dependent; β-thalassemia major patients - 5, who were on regular blood transfusion; severe HbE-β-thalassemia patients - 6, who had 6-12 transfusion requirements annually) were included. Informed consent was taken from the patients and parents of pediatric patients and the Ethical Committee of the National Institute of Immunohematology approved these studies.
In-vivo studies
Hydroxyurea (Cytodrox, Cipla Ltd, Mumbai) was started at a dose of 10-15 mg/kg/day [9],[10] after initial blood collection for in-vitro studies, hematological investigations like complete blood count, hemoglobin analysis on the Variant Hemoglobin Testing System (BioRad Laboratories, Inc., Hercules, CA, USA) and quantitative estimation of fetal cells (F-cells) by flow cytometry using a monoclonal HbF antibody (BD (Becton Dickinson and Company) Immunocytometry Systems, San Jose, CA, USA). [12] Molecular analysis such as confirmation of the HbS (sickle hemoglobin) and HbE mutations and characterization of β-thalassemia mutations were carried out as described earlier. [13] Xmn I polymorphism analysis was carried out by PCR (polymerase chain reaction) and restriction enzyme digestion. [14] Absolute quantification of gamma (γ)-globin mRNA transcripts was carried out by real time PCR using Taqman probes and the 7900 High Temperature Fast Real-Time PCR System (Applied Biosystems, NJ, USA) as described earlier. [15] The expression of the housekeeping gene β-actin was used as control in each sample. Patients were assessed clinically and laboratory investigations carried out monthly for 24 months to evaluate their status after starting hydroxyurea therapy. The numbers of remaining capsules of hydroxyurea were counted during each follow-up to evaluate therapeutic compliance and the hematological status was monitored to rule-out cytopenia. At the end of 24 months the patients' response to hydroxyurea therapy was evaluated by the reduction in clinical severity of the disease in case of sickle cell disease patients and reduction or cessation of transfusion requirements in β-thalassemia and HbE-β-thalassemia patients. After starting hydroxyurea therapy, the β-thalassemia patients were transfused only if their hemoglobin dropped below 7.5 g/dl. Partial responders of hydroxyurea therapy among the β-thalassemia were those patients whose transfusion requirement reduced by 50% whereas non-responders were those patients who did not show any reduction in transfusion requirements after 1 year of hydroxyurea therapy.
In-vitro studies
In-vitro studies were performed using the two phase liquid culture technique. [16],[17] Mononuclear cells, collected from peripheral blood of all patients before starting hydroxyurea were cultured in phase-I medium containing serum free StemSpan medium (StemCell Technologies Inc.), 50 ng/ml of stem cell factor (SCF), 25 ng/ml of interleukin-3 (IL3), 0.01% bovine serum albumin (BSA) and Cyclosporin A (1 μg/ml) (Sigma-Aldrich Co. USA) for 7 days at 37°C and 5% CO 2 . Non-adherent cells after day-7 were further cultured in phase-II medium consisting of StemSpan medium, 2 U/ml of human recombinant erythropoietin, 50 ng/ml SCF and 10 -7 M Dexamethazone (Sigma-Aldrich Co. USA). The culture was then bifurcated with a cell concentration of less than 1 × 10 6 /ml and hydroxyurea (1.5 mM/1 × 10 6 cells) was added to one of the cultures between days 6 and 8 of phase-II. After 10-12 days the cells were collected for F-cells estimation and γ-mRNA expression as described above.
Flow cytometry
For F-cells estimation, the cultured cells were washed twice and blocked with 2% BSA and stained with cell surface markers like anti-human-CD45 (leukocyte common antigen) tagged with perCP (Peridinin Chlorophyll Protein Complex) (10 μl) and anti-human-CD71 (transferrin receptor) tagged with Phycoerythrin (10 μl) and incubated in the dark for 30 min. After washing, the fluorescently labeled cells were fixed, permeabilized and stained with anti-HbF-FITC (Fluorescein isothiocyanate) as mentioned earlier. [12] Cultured cells, which were CD45 negative were gated (10,000 cells) and used for flow cytometric analysis to quantitate the number of F-cells.
Statistical analysis
Student t-test was used to compare the findings of in-vitro studies with and without hydroxyurea and of patients before and after hydroxyurea therapy. A P < 0.01 was considered statistically significant.
| Results | | |
Clinical and hematological response to hydroxyurea therapy in-vivo
After hydroxyurea therapy, 8 patients, which include 6 sickle cell anemia patients and 2 sickle-β-thalassemia patients had no further episodes of vaso-occlusive crisis, need for blood transfusions, infections, stroke or acute chest syndrome and they had a feeling of general well-being after therapy. The mean hematological parameters of these 8 patients also showed a significant increase in Hb, MCV (Mean corpuscular volume), HbF, and F-cells (P < 0.001) [Table 1] after hydroxyurea therapy. The γ-mRNA expression carried out in 2 sickle-β-thalassemia patients also showed a 390% and 255% increase after hydroxyurea therapy [Table 2].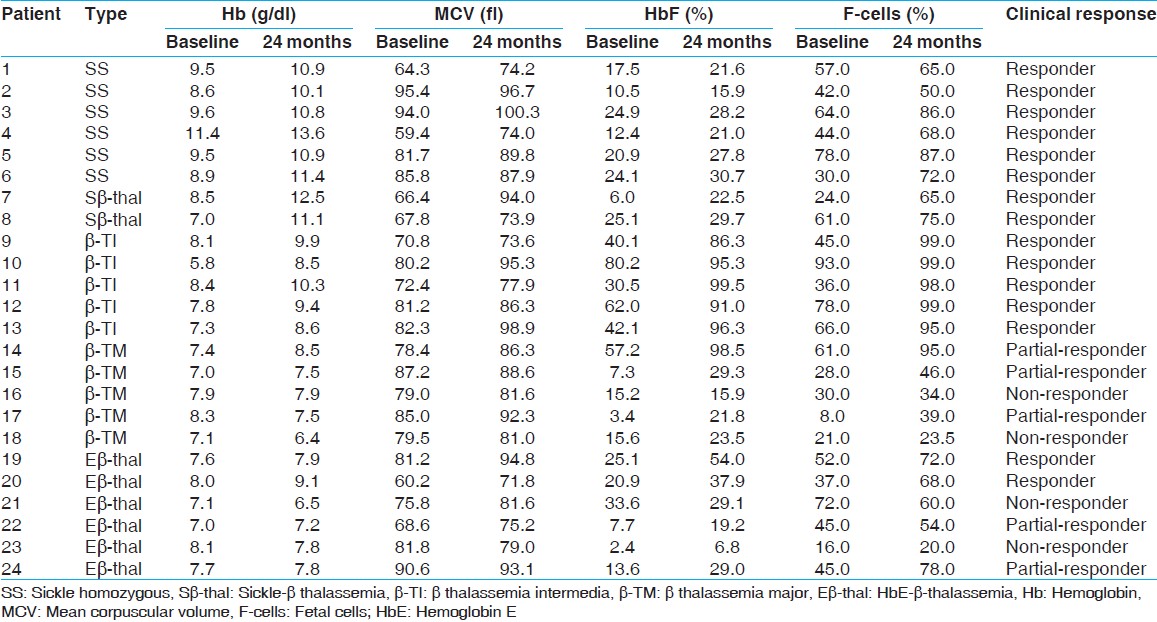 | Table 1: Hematological parameters of patients with sickle cell anemia, sickle-β-thalassemia, β-thalassemia intermedia, β-thalassemia major and HbE-β-thalassemia before and after hydroxyruea therapy
Click here to view |
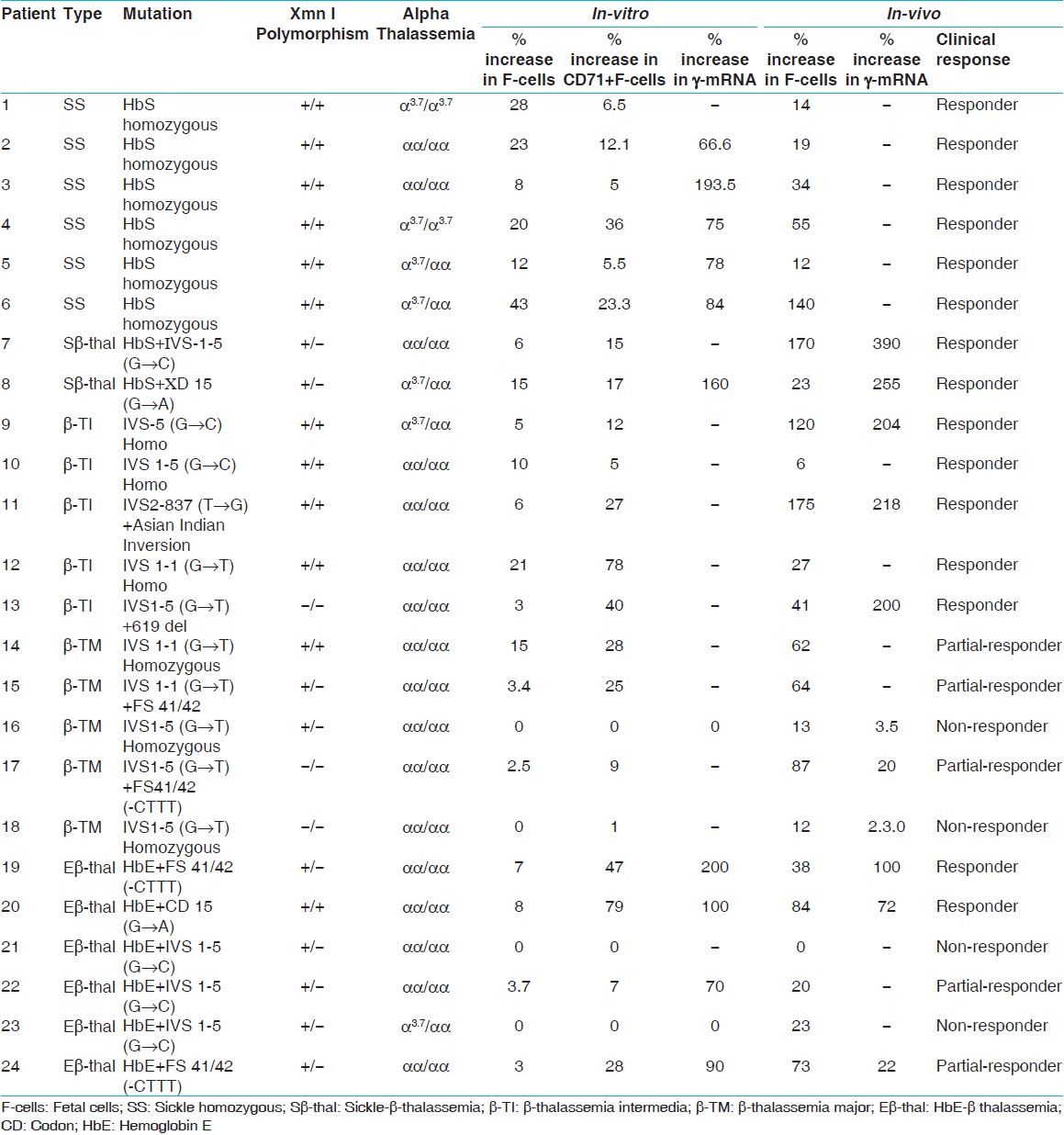 | Table 2: Comparison of the percent increase in F-cells, CD71+F-cells and g -mRNA expression of cultured erythroid cells (in-vitro) with the in-vivo percent increase in F - cells and mRNA expression in few patients before and after hydroxyurea therapy
Click here to view |
5 β-thalassemia intermedia patients who presented after 2 years of age and required blood transfusions every 20-25 days thereafter responded by not having any further transfusion requirements after 3-6 months of hydroxyurea therapy and maintained a Hb level of 8-10 g/dl. They showed a significant increase in mean Hb, MCV, HbF, and F-cells (P < 0.001) after hydroxyurea therapy [Table 1].
3 of the 5 β-thalassemia major patients, showed a reduction in their transfusion requirements from 12 to 15 transfusions per year to 7-8 transfusions per year. Their Hb levels remained between 7 and 8 g/dl for 45-60 days without transfusion. An increase was seen in HbF and F-cells after hydroxyurea therapy. However, 2 non-responders failed to show any change in their transfusion requirements or hematological parameters after 1 year of therapy. The mean increase in HbF and F-cells was not sufficient to maintain a stable Hb level without transfusion. A total of 2 patients (1 partial-responder and 1 non-responder) had two episodes of neutropenia because of which the hydroxyurea was discontinued until the counts recovered. No other side-effect was observed.
2 of the 6 HbE-β-thalassemia patients requiring 12-15 transfusions per year, responded to hydroxyurea therapy with discontinuation of transfusion requirements and a significant increase in mean Hb and MCV (P < 0.01) and HbF and F-cells (P < 0.001). Two patients were partial-responders with increase only in HbF and F-cells and two patients were non-responders [Table 1].[Table 3] shows the mean percent increase in F-cells, CD71 + F-cells and γ-mRNA expression seen among the responders, partial-responders, and non-responders of hydroxyurea therapy both in-vitro and in-vivo. The difference in the percent increase in F-cells seen in responders and partial-responders was not statistically significant (P < 0.1). However, the difference in the increase in γ-mRNA expression seen in responders and partial-responders was statistically significant (P < 0.01). | Table 3: Percent increase in F-cells, CD71 +F-cells and γ-mRNA expression in-vitro and in-vivo among the responders, partial-responders and non-responders of hydroxyurea therapy
Click here to view |
Response to hydroxyurea therapy in-vitro
The erythroid cells of the patients were cultured for an average of 18.5 ± 4 days. Erythroblasts increased dramatically from day 1 to 4 with a decrease in cell volume from days 5 to 7. Orthochromatic normoblasts representing the late erythroblastic cells stage rarely lead to enucleation under the culture conditions. Hydroxyurea added on day 7 ± 1 in one of the duplicate sets of culture to evaluate the F-cells response was titrated and a dose of 1.5 mM/1 × 10 6 cells was found to be optimum for the cells. A concentration above 1.5 mM reduced the cell number and the expression of CD71. [Figure 1] shows the flow cytometric data of cultured cells showing co-positivity for F-cells and CD71 before and after addition of hydroxyurea.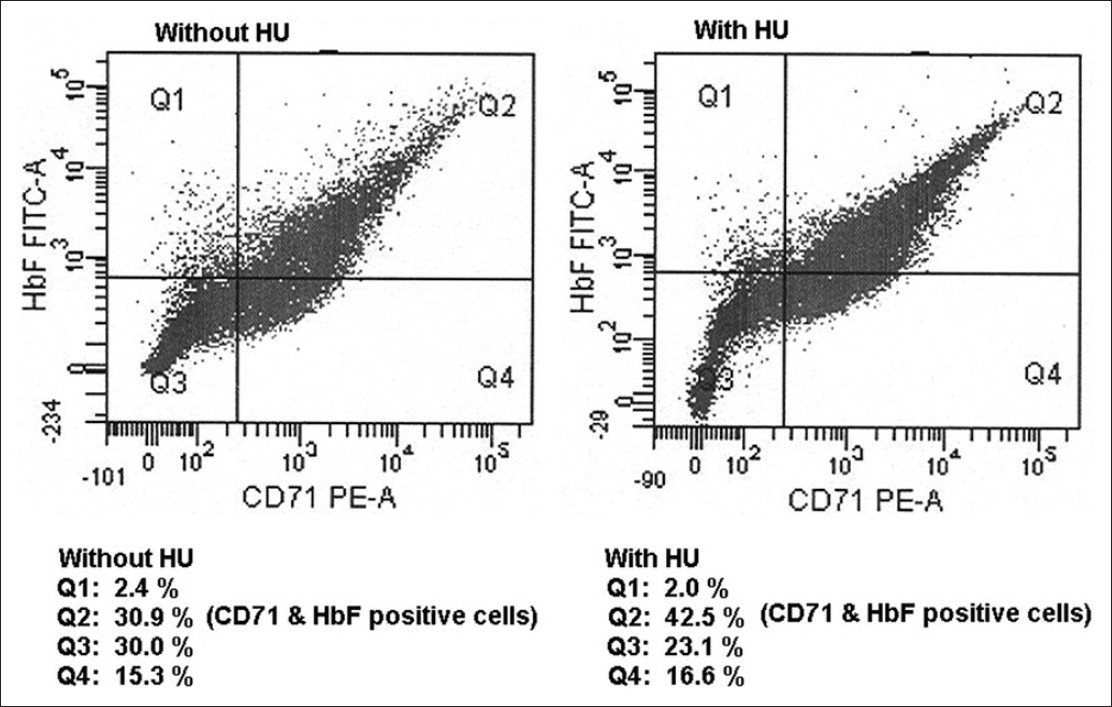 | Figure 1: Flow cytometric estimation of CD71 and fetal cells on cultured erythroid cells before and after addition of hydroxyurea
Click here to view |
The percent increase in F-cells, F-cells + CD71 and γ-mRNA expression of individual patient's cultured erythroid cells after treating with hydroxyurea is shown in [Table 2]. The difference in F-cells + CD71 and γ-mRNA expression seen among the responders and partial-responders was not statistically significant [Table 3]. 4 non-responders in-vivo, did not show any increase in F-cells in their cultured cells treated with hydroxyurea. Only 1 of the 4 non-responders (β-thalassemia major patient) showed an increase in F-cells + CD71 by 1% after hydroxyurea treatment, whereas the other 3 patients did not show any increase. No increase was found in the γ-mRNA expression carried out in 2 of the 4 non-responders' cultured erythroid cells after hydroxyurea treatment.
| Discussion | | |
Many pharmacological agents have been studied to enhance the production of HbF in various in-vivo and in-vitro systems. Use of these drugs on non-human primates are a pre-requisite for clinical trials in patients, however, they are too expensive and time consuming for large scale screening of these compounds. Hence, attempts have been made to mimic the in-vivo condition in-vitro by various experimental systems using either immortalized cell lines or isolation and culture of erythroid progenitor cells from the bone marrow or peripheral blood. Fibach et al. in 1989 established primary erythroid cultures by isolating erythroid progenitor cells from bone marrow, cord blood or peripheral blood, which when grown in-vitro resembles more closely the in-vivo situation. [18] Fibach in 1998 studied many pharmacological inducers of HbF like hydroxyurea and 5-azacytidine using peripheral blood of sickle cell anemia patients with this technique. [16] Treatment with hydroxyurea resulted in reduction in cell number, increase in MCV, MCH and HbF. It was further suggested that this technique could be used to predict the responders of hydroxyurea.
In 2002, Wojda et al. found that HbA and HbF content were regulated with the rate of proliferation during adult erythropoiesis and that there was no evidence for a HbF dominant population or switching during differentiation in adult cells. [19] Wojda et al. in 2003 showed significant SCF mediated increase in HbF using progenitor cells derived from donor cells. [20] Mithramycin and rapamycin were shown to induce HbF production by up regulating γ-globin mRNA production in normal and thalassemia human erythroid precursor cells using the two phase liquid culture technique. [21],[22] Human erythroid progenitor cells isolated from 18 HbE-β-thalassemia patients were treated with 30 μmol/l hydroxyurea in-vitro and a 30% increase in HbF was found in 4 patients, 20-30% increase in 9 patients and less than 20% in 5 patients. [23] Jiang et al. in 2006 studied the expression of 5 protein coding genes, ALDH (aldehyde dehydrogenase) 8A1, HBS1 L, cMYB (myeloblastosis), AHI1 (Abelson helper integration site 1) and PDE7B (Phosphodiesterase 7B) on chromosome 6q23 by using erythroid cell culture technique and revealed a clear correlation of elevated HbF with low levels of cMYB expression and its involvement in regulation of HbF. [17] Rigano et al. in 2010 studied 17 β-thalassemia intermedia patients in-vivo on hydroxyurea and confirmed that the erythroid cultures obtained from patients during treatment reproduced the observed in-vivo response. [24]
In this study, the response to hydroxyurea is compared in-vivo as well as in-vitro. Our earlier study on the use of hydroxyurea among sickle cell disease patients in India has shown the drug to be very effective in reducing clinical crisis, [11] however, only 60% of the β-thalassemia intermedia and 50% of HbE-β-thalassemia patients fully benefited from the drug. [12],[25] The in-vitro study on the effect of hydroxyurea on the cultured erythrocytes of patients with different Hemoglobinopathies gives an idea about the parameters that may predict the response to hydroxyurea therapy among these patients.
Hydroxyurea was found to be beneficial both among adults and children with severe form of sickle cell disease [8],[26],[27],[28] and was further used in the treatment of patients with β-thalassemia and HbE-β-thalassemia where a good response was seen among a few β-thalassemia intermedia and HbE-β-thalassemia patients in whom many genetic and epigenetic factors are believed to play a role in predicting the response to hydroxyurea therapy. [10],[11],[25],[29],[30],[31],[32] Watanapokasin et al. 2005 showed that 13 β-thalassemia/HbE-β-thalassemia patients treated with hydroxyurea in-vivo had HbF synthesis and γ-globin-mRNA that correlates with the in-vitro results suggesting that in-vitro testing may predict the in-vivo response to hydroxyurea. [23] Bianchi et al. 2007 used 3 inducers of HbF from the biological materials, which included angelicin linear psoralens, resveratrol and rapamycin. A good correlation was found between in-vitro and in-vivo response to HbF production. [33]
In our study, a significantly higher expression of γ-mRNA (P < 0.01) was seen among the cultures of responders treated with hydroxyurea as compared to γ-mRNA expression seen among partial-responders. The non-responders did not show any increase in F-cells or γ-mRNA expression in-vitro, however, they showed a 16.0 ± 6.1% increase in F-cells but only a 2.9 ± 0.8% increase in γ-mRNA expression in-vivo after therapy. Thus γ-mRNA expression was a better indicator of the response to hydroxyurea therapy both in-vivo and in-vitro as compared to the F-cells level as the F-cells showed an increase in most of the patients and their cultured erythrocytes treated with hydroxyurea, however, the percentage increase in F-cells levels differed among the responders and the non-responders.
This method of erythroid cell culture may be useful to compare the in-vivo response of hydroxyurea with in-vitro using γ-mRNA expression and the number of F-cells. However, a much larger study is required to confirm these results. This culture system could be further used to identify other pharmacological inducers of HbF production, which could be useful for non-responders of hydroxyurea and to study expression of other genes that may be involved in HbF production.
| References | | |
| 1. | Weatherall DJ, Clegg JB. Distribution and population genetics of the thalassemias. In: Weatherall DJ, Clegg JB, editors. The Thalassemia Syndromes. 4 th ed. Oxford University Press: Blackwell Scientific Publications; 2001. p. 241-246. 
|
| 2. | Mohanty D, Mukherjee MB. Sickle cell disease in India. Curr Opin Hematol 2002;9:117-22.
|
| 3. | Sukumaran PK. Abnormal hemoglobins in India. In: Sen NN, editor. Trends in Haematology, JB Chatterjea Memorial Volume. Calcutta: JB Chatterjea Memorial Committee; 1975. p. 227-8.
|
| 4. | Madan N, Sharma S, Sood SK, Colah R, Bhatia LH. Frequency of β-thalassemia trait and other hemoglobinopathies in northern and western India. Indian J Hum Genet 2010;16:16-25.
[PUBMED]  |
| 5. | Agarwal MB. The burden of haemoglobinopathies in India - Time to wake up? J Assoc Physicians India 2005;53:1017-8.
|
| 6. | Mohanty D, Colah R, Gorakshakar AC editors. Report of the Jai Vigyan S and T Mission Project on Community Control of Thalassemia Syndromes-Awareness, Screening, Genetic Counseling and Prevention. In: Mohanty D, Colah R, Gorakshakar AC, editors. New Delhi: Indian Council of Medical Research; 2008.
|
| 7. | Mukherjee MB, Lu CY, Ducrocq R, Gangakhedkar RR, Colah RB, Kadam MD, et al. Effect of alpha-thalassemia on sickle-cell anemia linked to the Arab-Indian haplotype in India. Am J Hematol 1997;55:104-9.
|
| 8. | Italia K, Jain D, Gattani S, Jijina F, Nadkarni A, Sawant P, et al. Hydroxyurea in sickle cell disease - A study of clinico-pharmacological efficacy in the Indian haplotype. Blood Cells Mol Dis 2009;42:25-31.
|
| 9. | Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332:1317-22.
|
| 10. | Alebouyeh M, Moussavi F, Haddad-Deylami H, Vossough P. Hydroxyurea in the treatment of major beta-thalassemia and importance of genetic screening. Ann Hematol 2004;83:430-3.
|
| 11. | Italia KY, Jijina FJ, Merchant R, Panjwani S, Nadkarni AH, Sawant PM, et al. Response to hydroxyurea in beta thalassemia major and intermedia: Experience in western India. Clin Chim Acta 2009;407:10-5.
|
| 12. | Italia KY, Colah R, Mohanty D. Evaluation of F cells in sickle cell disorders by flow cytometry - Comparison with the Kleihauer-Betke's slide method. Int J Lab Hematol 2007;29:409-14.
|
| 13. | Colah RB, Gorakshakar AC, Lu CY, Nadkarni AH, Desai SN, Pawar AR, et al. Application of covalent reverse dot blot hybridization for rapid prenatal diagnosis of the common Indian thalassemia syndromes Indian J Hematol Blood Transfus 1997;15:10-3.
|
| 14. | Old JM. DNA based diagnosis of the hemoglobin disorders. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Disorders of Hemoglobin - Genetics, Pathophysiology and Clinical Management. Cambridge: Cambridge University Press; 2001. p. 941-8.
|
| 15. | Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 2000;25:169-93.
|
| 16. | Fibach E. Techniques for studying stimulation of fetal hemoglobin production in human erythroid cultures. Hemoglobin 1998;22:445-58.
|
| 17. | Jiang J, Best S, Menzel S, Silver N, Lai MI, Surdulescu GL, et al. cMYB is involved in the regulation of fetal hemoglobin production in adults. Blood 2006;108:1077-83.
|
| 18. | Fibach E, Manor D, Oppenheim A, Rachmilewitz EA. Proliferation and maturation of human erythroid progenitors in liquid culture. Blood 1989;73:100-3.
|
| 19. | Wojda U, Noel P, Miller JL. Fetal and adult hemoglobin production during adult erythropoiesis: Coordinate expression correlates with cell proliferation. Blood 2002;99:3005-13.
|
| 20. | Wojda U, Leigh KR, Njoroge JM, Jackson KA, Natarajan B, Stitely M, et al. Fetal hemoglobin modulation during human erythropoiesis: Stem cell factor has "late" effects related to the expression pattern of CD117. Blood 2003;101:492-7.
|
| 21. | Fibach E, Bianchi N, Borgatti M, Prus E, Gambari R. Mithramycin induces fetal hemoglobin production in normal and thalassemic human erythroid precursor cells. Blood 2003;102:1276-81.
|
| 22. | Mischiati C, Sereni A, Lampronti I, Bianchi N, Borgatti M, Prus E, et al. Rapamycin-mediated induction of gamma-globin mRNA accumulation in human erythroid cells. Br J Haematol 2004;126:612-21.
|
| 23. | Watanapokasin R, Sanmund D, Winichagoon P, Muta K, Fucharoen S. Hydroxyurea responses and fetal hemoglobin induction in beta-thalassemia/HbE patients' peripheral blood erythroid cell culture. Ann Hematol 2006;85:164-9.
|
| 24. | Rigano P, Pecoraro A, Calzolari R, Troia A, Acuto S, Renda D, et al. Desensitization to hydroxycarbamide following long-term treatment of thalassaemia intermedia as observed in vivo and in primary erythroid cultures from treated patients. Br J Haematol 2010;151:509-15.
|
| 25. | Italia KY, Jijina FF, Merchant R, Panjwani S, Nadkarni AH, Sawant PM, et al. Effect of hydroxyurea on the transfusion requirements in patients with severe HbE-beta-thalassaemia: A genotypic and phenotypic study. J Clin Pathol 2010;63:147-50.
|
| 26. | Charache S, Dover GJ, Moore RD, Eckert S, Ballas SK, Koshy M, et al. Hydroxyurea: Effects on hemoglobin F production in patients with sickle cell anemia. Blood 1992;79:2555-65.
|
| 27. | Kinney TR, Helms RW, O'Branski EE, Ohene-Frempong K, Wang W, Daeschner C, et al. Safety of hydroxyurea in children with sickle cell anemia: Results of the HUG-KIDS study, a phase I/II trial. Pediatric Hydroxyurea Group. Blood 1999;94:1550-4.
|
| 28. | Hankins JS, Ware RE, Rogers ZR, Wynn LW, Lane PA, Scott JP, et al. Long-term hydroxyurea therapy for infants with sickle cell anemia: The HUSOFT extension study. Blood 2005;106:2269-75.
|
| 29. | Bradai M, Abad MT, Pissard S, Lamraoui F, Skopinski L, de Montalembert M. Hydroxyurea can eliminate transfusion requirements in children with severe beta-thalassemia. Blood 2003;102:1529-30.
|
| 30. | Yavarian M, Karimi M, Bakker E, Harteveld CL, Giordano PC. Response to hydroxyurea treatment in Iranian transfusion-dependent beta-thalassemia patients. Haematologica 2004;89:1172-8.
|
| 31. | Fucharoen S, Siritanaratkul N, Winichagoon P, Chowthaworn J, Siriboon W, Muangsup W, et al. Hydroxyurea increases hemoglobin F levels and improves the effectiveness of erythropoiesis in beta-thalassemia/hemoglobin E disease. Blood 1996;87:887-92.
|
| 32. | Singer ST, Kuypers FA, Olivieri NF, Weatherall DJ, Mignacca R, Coates TD, et al. Single and combination drug therapy for fetal hemoglobin augmentation in hemoglobin E-beta 0-thalassemia: Considerations for treatment. Ann N Y Acad Sci 2005;1054:250-6.
|
| 33. | Bianchi N, Zuccato C, Lampronti I, Borgatti M, Gambari R. Fetal hemoglobin inducers from the natural world: A novel approach for identification of drugs for the treatment of β-thalassemia and sickle-cell anemia. Evid Based Complement Altern Med eCAM 2009; 6:141-151.
|
[Figure 1]
[Table 1], [Table 2], [Table 3]
|










