|
 
 |
|
ORIGINAL ARTICLE |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 3 | Page : 337-341 |
| |
Analysis of hemoglobin electrophoresis results and physicians investigative practices in Saudi Arabia
Syed Riaz Mehdi1, Badr Abdullah Al Dahmash2
1 Department of Pathology, Era's Lucknow Medical College, Lucknow, Uttar Pradesh, India
2 College of Applied Medical Sciences, King Saud University, Riyadh, Saudi Arabia
| Date of Web Publication | 30-Oct-2013 |
Correspondence Address:
Syed Riaz Mehdi
505, Dilkash Apartment, 3-River Bank Colony, Lucknow - 226 018, Uttar Pradesh
India
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.120829

 Abstract Abstract | | |
Background and Objectives: Riyadh and central province falls in a moderate prevalent zone of hemoglobinopathies in Saudi Arabia. However, it has been observed that the physicians working in Saudi Arabia invariably advise all cases of anemia for hemoglobin electrophoresis (HE). The present work was carried out to study the yield of the HE in Riyadh and the investigative practices of the physicians advising HE.
Settings and Design: The study was carried out in the hospitals of King Saud University from 2009 to 2011 in order to assess the yield of HE in referred cases of clinical anemia.
Materials and Methods: A total of 1073 cases divided in two groups of males and females had undergone complete blood count and red blood cell morphology. Cellulose acetate HE was performed and all the positive results were reconfirmed on the high performance liquid chromatography (HPLC). The results were analyzed for the type of hemoglobinopathies. For statistical analysis Statistical Package for Social Sciences 15 version (SPSS Inc., Chicago, IL, USA) was used.
Results: A total of 405 males and 668 females blood samples were included in the present study. 116 (28.5%) males and 167 (25%) females showed an abnormal pattern on HE. The incidence of beta thalassemia trait was higher in females while sickle cell trait was predominantly seen in males. Red cell indices were reduced considerably in thalassemias, but were unaffected in sickle cell disorders, except those which had concurrent alpha trait. The total yield of HE was 26.6% which was much less than expected.
Conclusion: The physicians are advised to rule out iron deficiency and other common causes of anemia before investigating the cases for hemoglobinopathies, which employs time consuming and expensive tests of HE and HPLC.
Keywords: Electrophoresis, hemoglobin, physician′s practices, Saudi Arabia
How to cite this article:
Mehdi SR, Al Dahmash BA. Analysis of hemoglobin electrophoresis results and physicians investigative practices in Saudi Arabia. Indian J Hum Genet 2013;19:337-41 |
How to cite this URL:
Mehdi SR, Al Dahmash BA. Analysis of hemoglobin electrophoresis results and physicians investigative practices in Saudi Arabia. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:337-41. Available from: http://www.ijhg.com/text.asp?2013/19/3/337/120829 |
| Introduction | |  |
Normal hemoglobin (Hb) A has got two α and two β chains. α gene clusters are present on chromosome 16 while β genes are present on chromosome 11. Hereditary Hb disorders are caused by structural abnormalities as a result of decreased production of either chain due to the deletion or mutation.
Hemoglobinopathies have been reported from every part of the world. [1],[2],[3] However, Mediterranean region, tropical and sub-tropical Africa, India, [4],[5] South East Asia and the middle East including Saudi Arabia [6],[7],[8] are the regions from where the maximum number of cases are reported.
In Saudi Arabia sickle cell anemia and thalassemias are prevalent predominantly in the eastern region, western and southern regions. [9],[10],[11],[12],[13] First cousin and other consanguineous marriages is the most important cause of higher prevalence of hemoglobinopathies in these regions. [12]
Thalassemias were first reported from the Eastern region by Lehman et al. in 1963. [14] Sickle cell heterozygotes and homozygotes are also more prevalent from the same region. Subsequently several studies on the prevalence and genetic structure of hemoglobinopathies have been carried out in the kingdom. [6],[7],[8],[15],[16],[17],[18],[19],[20] The overall prevalence of hemoglobinopathies in Saudi Arabia is definitely higher than the Europe and Americas. [1],[2],[3],[21] Central region and Riyadh fall in the low prevalent zones for hemoglobinopathies of Saudi Arabia. [6],[22]
In 2004 through a royal decree premarital screening of the couples for thalassemia and sickle cell anemia was made obligatory in Saudi Arabia. [22] If prospective husband and wife came out to be the carriers of thalassemia or sickle cell or either of the two is homozygote for the disease the couple is referred to the counseling centers and is advised to abstain from marriage.
Neonatal screening for hemoglobinopathies is a part of a national program adopted by Saudi Arabia. [9],[22]
A scientific advisory committee for Hereditary Blood Disease Centers (HBDC) for every region of Saudi Arabia was formed in 2008. [22]
These measures have considerably reduced the incidence of hemoglobinopathiesin the Kingdom in the last few years.
The present study was designed to analyze retrospectively the results of hemoglobin electrophoresis (HE) carried out in Riyadh region and to assess the justification of HE advised by the physicians managing anemia.
| Materials and Methods | | |
The present study was carried out in the hematology laboratory of the college of health sciences and the hospitals of the King Saud University Riyadh, Saudi Arabia from 2009 to 2011.
A total of 405 males and 668 females (in all 1073) from adult and pediatric age groups presenting with clinical anemia were referred for HE. 2 ml blood sample from every patient was collected in an ethylenediaminetetraacetic acid vacutainer for estimation of hemoglobin (Hb), hematocrit (Hct), total red blood cell count, total leukocyte count, platelet count, mean cell volume (MCV), mean cell hemoglobin (MCH) and mean cell hemoglobin concentration (MCHC). The tests were performed on a Beckman Coulter (USA) hematology analyzer. MCV < 78 fl was considered microcytosis and MCH < 27 pg and MCHC < 32 g/dl were considered hypochromic. RBC morphology was studied on Wright's stained peripheral blood smears (PBS). Reticulocyte count was carried out on methylene blue stained smears.
Hemolysate was prepared from washed cells treated by cold distilled water, which was subjected to HE at alkaline pH of 8.6 in cellulose acetate medium by exprime 72 methodology provided by Amplimedical corporation, Italy (Associate of Roche, USA). All Hb variants discovered on HE were reconfirmed on high performance liquid chromatography (HPLC). Hb F was also estimated by alkali denaturation technique of Betke et al. [23]
Level of Hb A2 > 3.5% was considered the cut off for β thalassemia trait.
For α- and β-genotyping, genomic deoxyribonucleic acid was prepared from peripheral blood by the standard phenol chloroform extraction method.
Statistical analysis was performed by Statistical Package for Social Sciences (SPSS) version 15 software (SPSS Inc., Chicago, IL, USA) and an independent t-test was used for comparison of hematological parameters. Results are presented as mean values and standard deviation (SD). A P < 0.05 was considered statistically significant.
| Results | | |
1073 HE results were analyzed in the present study, out of which 668 were females and 405 males. The adults' age range was 14-61 years with a mean age of 30.97 ± 9.95 while in the children group of 577 the age range was 1-9 years with a mean of 4.44 ± 2.82.
289 (71.5%) males and 501 (75%) females had normal AA pattern on HE. 116 (28.5%) males and 167 (25%) of females had shown abnormal Hb pattern.
The significant finding was the predominance of female carriers of β thalassemia trait 91 (13.6%) and of males 43 (10.3%) with sickle cell trait in the study population. The males were also ahead of females in all other types of hemoglobinopathies.
Four cases (0.9%) of Hb E heterozygotes were detected in males and only 2 (0.2%) in females. Out of these six cases, only two patients were Filipinos while the rest four were Saudis. The D variant heterozygotes were found exclusively in males.
The number and percentage of each type of hemoglobinopathy in both groups of males and females is given in [Table 1].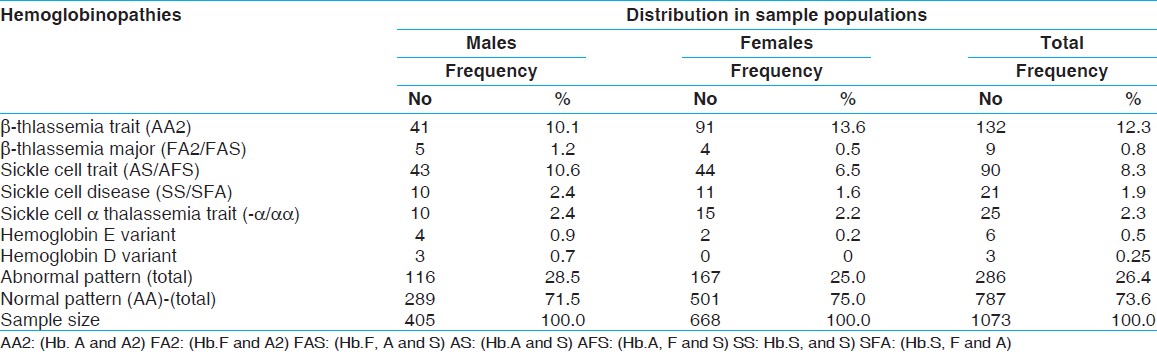 | Table 1: Sex wise distribution of Hemoglobinopaties in the study population
Click here to view |
The reticulocyte count was in the range of 3-4% in the positive cases. Hb and Hct were mildly low in cases of thalassemia and sickle cell trait while moderate to markedly low in cases of thalassemia major and sickle cell disease. The MCV and MCH were significantly low (P < 0.001) in cases of thalassemias presenting microcytic hypochromic picture on PBS, however, these values were within the normal limits in sickle cell disorders. The red cell count was increased in cases of thalassemias while it was not much affected in sickle cell disorders. The indices were lower in sickle cell α thalassemia trait (P < 0.05) [Table 2]. Two cases of Hb E heterozygote had low MCV and MCH.
| Discussion | | |
Extensive studies have been carried out in Saudi Arabia in the last two decades to study the prevalence and genotypes of different hereditary Hb disorders. Very high prevalence of sickle cell anemia, α and β thalassemias and interaction of genes have been reported from the eastern region of Saudi Arabia. [6],[7],[8],[15],[16],[17],[18],[19],[20],[22]
In the present study, we had analyzed the results of cases of anemia, which had undergone HE. Out of 1073 samples screened only 286 (26.3%) showed an abnormal pattern demonstrating a lower yield compared with reports from eastern and western regions. The results of the premarital screening and other population based studies have placed central region and Riyadh in a moderate prevalence zone for hemoglobinopathies. [6],[22]
The results of the blood screening carried out in different HBDC of Saudi Arabia in 2007 showed prevalence of 4.2% carriers and 0.26% of sickle cell disease while 3.22% carriers and 0.07% homozygotes of thalassemia at the national level.
In Riyadh, 2.08% carriers of sickle cell and 2.01% of thalassemia and 0.15% of the disease of both sickle and thalassemia were reported. [22] Sickle cell disorders comprise 74% of all hemoglobinopathies in Saudi Arabia. [22]
The statistically significant sex based difference in the incidence of hemoglobinopathies was observed in β thalassemia trait and sickle cell trait. The incidence of β thalassemia trait was higher in females while sickle cell trait was higher in males. In accordance with the previous studies from this region, we too have found a significant difference in the red cell indices of thalassemias and sickle cell disorders. [7],[16],[17] Invariably the MCV, MCH and MCHC remain within normal limits in the cases of sickle cell carriers and disease while they are reduced in thalassemias. α gene interaction in cases of sickle cell anemia leads to reduction of MCV and MCH. [16],[24],[25]
Since the present study is a hospital based analysis of the cases of clinical anemia referred for HE and not a general population based study, hence the number of positive cases are much higher compared with the prevalence reported earlier in the general population of Saudi Arabia. However, the higher percentage of positive results does reflect the higher prevalence of hemoglobinopathies in the general population. The findings correspond to the previous studies from Saudi Arabia and other Gulf states. [26],[27]
The Hb variant E and D, which are more prevalent in Southeast Asia [28] are rarely found among Saudis. Incidentally we had detected four cases of Hb E in Saudis. [18],[29] However, our results show relatively higher incidence of these rare Hb variants. The red cell indices were lower only in two cases of Hb E heterozygotes. The number of E and D cases being very few has not been displayed in the [Table 2].
The most common cause of microcytic hypochromic anemia is iron deficiency anemia (IDA). It is obligatory on physicians to first rule out IDA and then investigate for hemoglobinopathies, [26],[30] otherwise the overenthusiastic approach inflates the cost of treatment for the patient. In our study, only 26.6% cases of anemia yielded a positive result on HE.
The findings of our analysis could not be attributed to a particular ethnic group too since Riyadh is a metropolis of the cosmopolitan nature and here people from almost every region of Saudi Arabia reside.
| Conclusion | | |
Riyadh region is having a low incidence of hemoglobinopathies compared with the Eastern and Western regions of Saudi Arabia. It has been observed that physicians have a practice of advising HE invariably in every case of anemia before ruling out the common causes of anemia such as iron deficiency and chronic diseases, causing unnecessary burden on the laboratory resources and delay in initiating the proper management. The physicians are advised to follow the recommended protocol for investigating hemolytic anemia and must rule out IDA in cases of microcytic hypochromic blood picture before referring the patient for HE and HPLC.
| References | | |
| 1. | Embury SH, Dozy AM, Miller J, Davis JR Jr, Kleman KM, Preisler H, et al. Concurrent sickle-cell anemia and alpha-thalassemia: Effect on severity of anemia. N Engl J Med 1982;306:270-4. 
|
| 2. | Bleibel SA, Leonard RJ, Crawford JL, Kutlar A, Hendricks LK. Thalassemia Alpha; eMedicine>Hematology>Red Blood Cells Disorders. 2009.
|
| 3. | Lafferty JD, Barth DS, Sheridan BL, McFarlane AG, Halchuk LM, Crowther MA. Prevalence of thalassemia in patients with microcytosis referred for hemoglobinopathy investigation in Ontario: A prospective cohort study. Am J Clin Pathol 2007;127:192-6.
|
| 4. | Brittenham G, Lozoff B, Harris JW, Mayson SM, Miller A, Huisman TH. Sickle cell anemia and trait in southern India: Further studies. Am J Hematol 1979;6:107-23.
|
| 5. | Balgir RS. Hematological profile of twenty-nine tribal compound cases of hemoglobinopathies and G-6-PD deficiency in rural Orissa. Indian J Med Sci 2008;62:362-71.
[PUBMED]  |
| 6. | el-Hazmi MA, Warsy AS. Appraisal of sickle-cell and thalassaemia genes in Saudi Arabia. East Mediterr Health J 1999;5:1147-53.
|
| 7. | El-Hazmi MA, Lehmann H. Human haemoglobins and haemoglobinopathies in Arabia: Hb O Arab in Saudi Arabia. Acta Haematol 1980;63:268-73.
|
| 8. | Babiker MA, Taha SA. Two different patterns of sickle cell disease in children in Saudi Arabia. Ann Trop Paediatr 1982;2:179-81.
|
| 9. | Al-Awamy BH, Niazi GA, El Mouzan MI, AlTorki MT, Naeem MA. Newborn screening for sickle cell haemoglobinopathy and other inherited erythrocytic disprders in the eastern province of Saudi Arabia. Saudi Med J 1986;7:502-9.
|
| 10. | Al-Awamy BH, Niazi GA, el-Mouzan MI, Altorki MT, Naeem MA. Relationship of haemoglobin F and alpha thalassaemia to severity of sickle-cell anaemia in the Eastern Province of Saudi Arabia. Ann Trop Paediatr 1986;6:261-5.
|
| 11. | Acquaye JK, Omer A, Ganeshaguru K, Sejeny SA, Hoffbrand AV. Non-benign sickle cell anaemia in western Saudi Arabia. Br J Haematol 1985;60:99-108.
|
| 12. | El-Hazmi MA. Incidence and frequency of hemoglobinopathies and thalassemia in the North West sector Arabia. Saudi Med J 1985;6:149-62.
|
| 13. | el-Hazmi MA, Warsy AS. On the nature of sickle cell disease in the south-western province of Saudi Arabia. Acta Haematol 1986;76:212-6.
|
| 14. | Lehmann H, Maranjian G, Mourant AE. Distribution of sickle-cell hemoglobin in Saudi Arabia. Nature 1963;198:492-3.
|
| 15. | Al-Qurashi MM, El-Mouzan MI, Al-Herbish AS, Al-Salloum AA, Al-Omar AA. The prevalence of sickle cell disease in Saudi children and adolescents. A community-based survey. Saudi Med J 2008;29:1480-3.
|
| 16. | el-Hazmi MA. Studies on sickle cell heterozygotes in Saudi Arabia - Interaction with alpha-thalassaemia. Acta Haematol 1986;75:100-4.
|
| 17. | el-Hazmi MA. Haemoglobinopathies, thalassaemias and enzymopathies in Saudi Arabia: The present status. Acta Haematol 1987;78:130-4.
|
| 18. | El-Hazmi MA. The distribution and nature of hemoglobinopathies in Arabia: Hemoglobin variants in human population. In: Winter WP, editor. Boca Raton, Florida: CRC Press Inc; 1987. p. 65-77.
|
| 19. | el-Hazmi MA. Genetic red cell disorders in Saudi Arabia: A multifaceted problem. Hemoglobin 1994;18:257-68.
|
| 20. | Al-Awamy BH. Thalassemia syndromes in Saudi Arabia. Meta-analysis of local studies. Saudi Med J 2000;21:8-17.
|
| 21. | Brozoviæ M, Anionwu E. Sickle cell disease in Britain. J Clin Pathol 1984;37:1321-6.
|
| 22. | Alhamdan NA, Almazrou YY, Alswaidi FM, Choudhry AJ. Premarital screening for thalassemia and sickle cell disease in Saudi Arabia. Genet Med 2007;9:372-7.
|
| 23. | Betke K, Marti HR, Schlicht I. Estimation of small percentages of foetal haemoglobin. Nature 1959;184 Suppl 24:1877-8.
|
| 24. | Higgs DR, Aldridge BE, Lamb J, Clegg JB, Weatherall DJ, Hayes RJ, et al. The interaction of alpha-thalassemia and homozygous sickle-cell disease. N Engl J Med 1982;306:1441-6.
|
| 25. | Embury SH, Clark MR, Monroy G, Mohandas N. Concurrent sickle cell anemia and alpha-thalassemia. Effect on pathological properties of sickle erythrocytes. J Clin Invest 1984;73:116-23.
|
| 26. | Marouf R, D'souza TM, Adekile AD. Hemoglobin electrophoresis and hemoglobinopathies in Kuwait. Med Princ Pract 2002;11:38-41.
|
| 27. | Daar S, Hussain HM, Gravell D, Nagel RL, Krishnamoorthy R. Genetic epidemiology of HbS in Oman: Multicentric origin for the betaS gene. Am J Hematol 2000;64:39-46.
|
| 28. | Katsanis E, Luke KH, Hsu E, Yates JR. Hemoglobin E: A common hemoglobinopathy among children of Southeast Asian origin. CMAJ 1987;137:39-42.
|
| 29. | Owaidah TM, Al-Saleh MM, Al-Hellani AM. Hemoglobin D/beta-thalassemia and beta-thalassemia major in a Saudi family. Saudi Med J 2005;26:674-7.
|
| 30. | England JM, Fraser PM. Differentiation of iron deficiency from thalassaemia trait by routine blood-count. Lancet 1973;1:449-52.
|
[Table 1], [Table 2]
|









