|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 3 | Page : 355-357 |
| |
Genetic analysis of a family with complete androgen insensitivity syndrome
Sunil Kumar Kota1, Kotni Gayatri2, Siva Krishna Kota3, Sruti Jammula4
1 Department of Endocrinology, Medwin Hospital, Hyderabad, Andhra Pradesh, India
2 Department of Obstetrics and Gynecology, Riyadh Care Hospital, Riyadh, Saudi Arabia
3 Department of Anaesthesia, Central Security Hospital, Riyadh, Saudi Arabia
4 Department of Pharmaceuties, Roland Institute of Pharmaceutical Sciences, Berhampur, Orissa, India
| Date of Web Publication | 30-Oct-2013 |
Correspondence Address:
Sunil Kumar Kota
Department of Endocrinology, Medwin Hospitals, Chiragh Ali Lane, Nambally, Hyderabad - 500 001, Andhra Pradesh
India
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.120820

 Abstract Abstract | | |
Androgen insensitivity causes impaired embryonic sex differentiation leading to developmental failure of normal male external genitalia in 46 XY genetic men. It results from diminished or absent biological actions of androgens, which is mediated by the androgen receptor (AR) in both the embryo and secondary sexual development. Mutations in the AR located on the X chromosome are responsible for the disease. Almost 70% of affected individuals inherit the mutation from their carrier mother. We hereby report a 10-year-old girl with all the characteristics of complete androgen insensitivity syndrome (CAIS). Similar scenario was observed in 3 maternal aunts, Sequencing of the AR gene in all the family members revealed C 2754 to T transition in exon 6. It was concluded that the C 2754 to T transition rendered the AR incapable of both ligand-binding and activating the transcription and was the cause of CAIS in the patient.
Keywords: Androgen insensitivity, androgen receptor, ligand-binding domain, mutation
How to cite this article:
Kota SK, Gayatri K, Kota SK, Jammula S. Genetic analysis of a family with complete androgen insensitivity syndrome. Indian J Hum Genet 2013;19:355-7 |
How to cite this URL:
Kota SK, Gayatri K, Kota SK, Jammula S. Genetic analysis of a family with complete androgen insensitivity syndrome. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:355-7. Available from: http://www.ijhg.com/text.asp?2013/19/3/355/120820 |
| Introduction | |  |
Androgens determine the expression of the male phenotype. Their activity is mediated by an androgen receptor (AR), which translocates to the nucleus and binds to the regulatory regions of specific chromosomal deoxyribonucleic acid (DNA) sequences (androgen response elements), to activate androgen dependent genes. [1]
The AR is encoded by the AR gene (Xq11-12). The gene is formed by 8 exons and 7 introns, spanning more than 90 kb and codes for a protein with four functional domains. These are (a) exon 1 encoding the N-terminal domain (NTD), which serves modulatory function (1586 bp); (b) exons 2 and 3 encode the DNA-binding domain (DBD) (152 bp and 117 bp); (c) the "hinge" region, which binds the NTD and DBD and consists of residues 628-669; (d) exons 4-8 encode the C-terminal ligand-binding domain (LBD), which vary from 131 bp to 288 bp in size. [2] The C-terminus of the LBD mediates the hormone dependent transcription activation function. Mutations in the AR LBD perturb the conformation of the helix, which is unable to efficiently bind the ligand dihydrotestosterone (DHT) and to transactivate known androgen response elements. [3]
Disorders of AR function due to mutations in the AR gene cause different forms of X-linked male pseudohermaphroditism, known as androgen insensitivity syndromes (AIS) affecting XY female individuals with the normal androgen production and metabolisms. AIS are estimated to be present in 1:20000-64000 male births. The presence of variable phenotypic expression allows the classification of AIS into complete androgen insensitivity syndrome (CAIS), partial androgen insensitivity and mild androgen insensitivity.
Approximately, 90% of molecular defects in the AR gene are single base mutations, mostly missense mutations. In addition to the point mutations, the AR gene contains regions of repetitive DNA sequences, trinucleotide repeat CAG and GGN that have been associated with a number of disorders, such as androgen insensitivity, male infertility and prostate cancer. [4] Here, we describe a familial case of CAIS presenting with similar mutations described in 3 generations.
| Case Report | | |
A 10-year-old girl, product of consanguineous marriage was referred for evaluation of palpable gonads in the inguinal region. She was born at term after an uneventful pregnancy by normal vaginal delivery with a birth weight of 2.6 kg and length of 48 cm. Bilateral inguinal gonads with short and blind ended vagina were detected at birth. She was followed-up over a period of 8 months when assignment of the female gender was decided; corrective procedure for inguinal gonads was planned, but did not occur. On physical examination, she had typical female external genitalia. Palpable gonads were found bilaterally in the inguinal region [Figure 1], pubertal stage was B4P5. The uterus was absent under pelvic sonograms. Hormonal evaluation revealed follicle stimulating hormone (5.4 mIU/mL; normal: 1.5-12.4), luteinizing hormone (21.2 mIU/mL; Normal: 1.7-8.6) and total testosterone (15 ng/mL; Normal: 2.86-8.1); chromosomal analysis showed diploid 46 XY karyotype. Testosterone and DHT synthesis defects were excluded by the normal rise of T and DHT after human chorionic gonadotrophin (HCG) stimulation (basal T: 15 ng/mL; basal DHT: 9.3 ng/mL, T/DHT ratio after HCG stimulation: 7:9). Gonadectomy was performed a few months later and histological analysis revealed bilateral testes with no evidence of malignancy. She subsequently underwent vaginoplasty and received therapy with estrogens. Three maternal aunts presented with primary amenorrhea with an adequate breast and pubic hair development and palpable gonads in the age group between 15 and 20 years and were treated with bilateral gonadectomy, vaginal reconstruction and estrogen supplementation.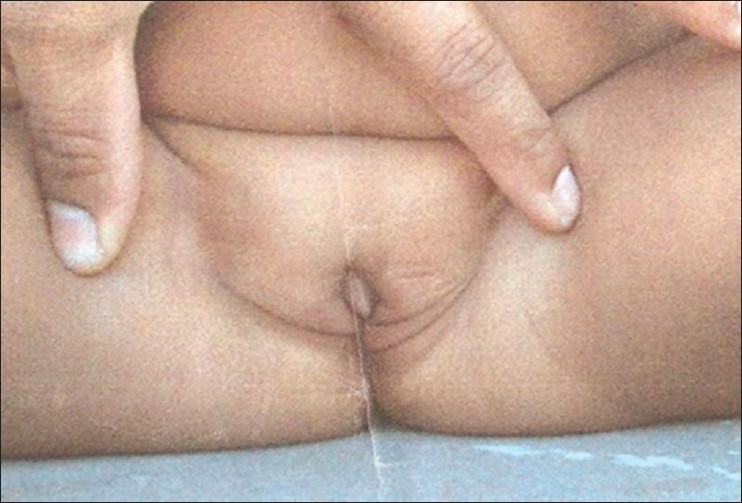 | Figure 1: Female appearing external genitalia with bilateral inguinal gonads
Click here to view |
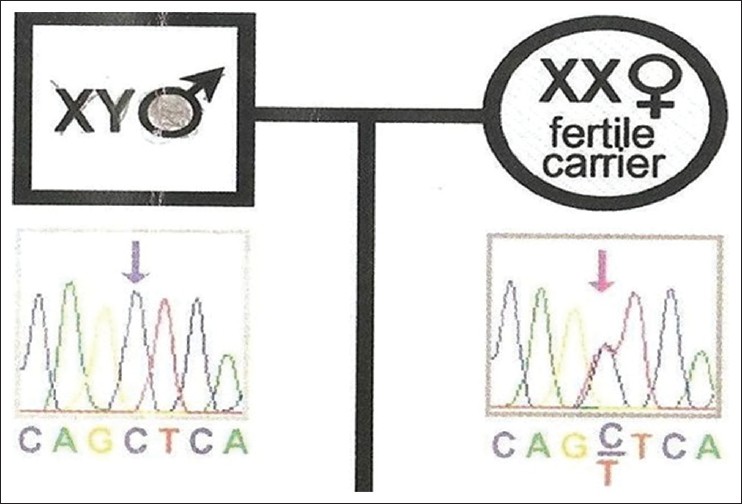
Peripheral blood samples were obtained from the girl and her maternal family members for molecular analysis. Genomic DNA was extracted with the use of polymerase chain reaction (PCR) amplification of AR exonic fragments 1-8 followed by direct sequencing analysis of the PCR products was performed. It revealed C 2754 to T transition in exon 6 [Figure 2]. The same mutation was confirmed in her mother and the 3 maternal aunts [Figure 3], maternal grandmother [Figure 4]. Her mother and maternal grandmother were fertile carriers with 46 XX karyotype. | Figure 2: Chromosomal and deoxyribonucleic acid analysis of the patient showing 46 XY karyotype with C 2754 to T transition of exon 6
Click here to view |
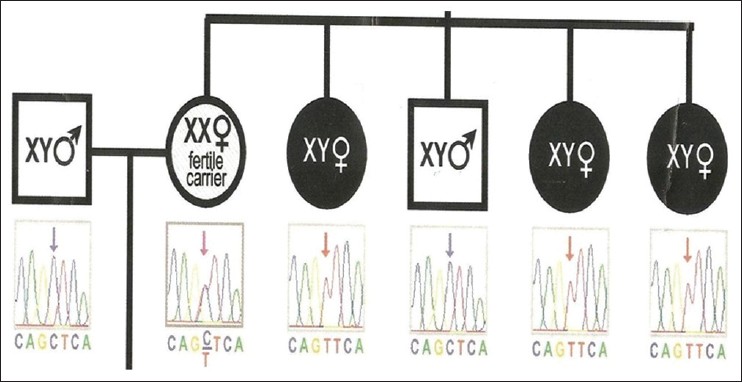 | Figure 3: Chromosomal and deoxyribonucleic acid analysis of the parents and maternal aunts. 3 affected maternal aunts showing 46 XY karyotype with C 2754 to T transition of exon 6. Mother is showing similar mutation with 46 XX karyotype, a fertile carrier
Click here to view |
 | Figure 4: Chromosomal and deoxyribonucleic acid analysis of the maternal grandmother showing 46 XX karyotype with C 2754 to T transition of exon 6
Click here to view |
| Discussion | | |
We report the familial occurrence of a mutation resulting from C to T transition. Six subjects in 3 generations carry the mutation. However, 4 were affected and 2 were 46 XX fertile carriers.
Missense mutations in AR protein may cause a spectrum of phenotypes. The phenotype variability appears to reflect the degree to which ligand-binding and receptor functions are disrupted by different substitutions. [5] In addition, genetic background also influences the resulting phenotype since a same mutation may cause different forms of AIS within a family. [6] The most frequent are missense mutations that are found within two important areas of the receptor protein: DBD and LDB domains. [7] Out of 193 mutations, 103 causing CAIS are located in the LBD, 51 in the N-terminal and 24 in the DBD [8] mutations of a single amino acid in the LBD of the AR can induce functional abnormalities in androgen binding, stabilization of active conformation or interaction with co-activators. In the AR C-terminal LBD, there is a clear predominance of missense mutations.
In our case mutation, which has been previously reported previously, alters a Gln codon to a termination codon (Q798X). [9] This results in the interruption of the amino acid sequence of the AR within the LBD between helices VII and VIII. The truncated form of the receptor is devoid of 123 amino acids at the carboxyl end, a major part of the LBD and the AT2 sequence responsible for the activation of the transcription. The previously reported lady had sertoli cell adenoma in both gonads, which was lacking in our patient. [9]
| Conclusion | | |
The characterization of mutations in the AR gene serves as a reliable tool for the diagnosis and molecular subclassification of AIS. In addition to the localization of the mutation within the gene sequence, the kind of amino acid substitution in mutation affects the resulting phenotype. Knowledge of the mutation in the AR and its functional consequences can provide useful information about the genotype-phenotype correlation, improving the management of cases of male pseudohermaphroditism pertinent to gender assignment, genital surgery and gonadectomy.
| References | | |
| 1. | Li BY, Liao XB, Fujito A, Thrasher JB, Shen FY, Xu PY. Dual androgen-response elements mediate androgen regulation of MMP-2 expression in prostate cancer cells. Asian J Androl 2007;9:41-50. 
|
| 2. | Hughes IA, Deeb A. Androgen resistance. Best Pract Res Clin Endocrinol Metab 2006;20:577-98.
|
| 3. | Adachi M, Takayanagi R, Tomura A, Imasaki K, Kato S, Goto K, et al. Androgen-insensitivity syndrome as a possible coactivator disease. N Engl J Med 2000;343:856-62.
|
| 4. | Rajender S, Singh L, Thangaraj K. Phenotypic heterogeneity of mutations in androgen receptor gene. Asian J Androl 2007;9:147-79.
|
| 5. | McPhaul MJ, Marcelli M, Tilley WD, Griffin JE, Wilson JD. Androgen resistance caused by mutations in the androgen receptor gene. FASEB J 1991;5:2910-5.
|
| 6. | Evans BA, Hughes IA, Bevan CL, Patterson MN, Gregory JW. Phenotypic diversity in siblings with partial androgen insensitivity syndrome. Arch Dis Child 1997;76:529-31.
|
| 7. | Brinkmann AO, Faber PW, van Rooij HC, Kuiper GG, Ris C, Klaassen P, et al. The human androgen receptor: Domain structure, genomic organization and regulation of expression. J Steroid Biochem 1989;34:307-10.
|
| 8. | Gottlieb B, Beitel LK, Wu JH, Trifiro M. The androgen receptor gene mutations database (ARDB):2004 update. Hum Mutat 2004;23:527-33.
|
| 9. | Ignacak M, Turek-Plewa J, Limon J, Trzeciak WH. A novel c.C2754 > T transition in the androgen receptor gene introduces the premature termination codon Q798X and results in a truncated form of the receptor. Gynecol Endocrinol 2004;19:178-81.
|
[Figure 1], [Figure 2], [Figure 3], [Figure 4]
|









