|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 3 | Page : 366-368 |
| |
A novel ABCB11 mutation in an Iranian girl with progressive familial intrahepatic cholestasis
Sassan Saber1, Reza Vazifehmand2, Iman Bagherizadeh3, Mahbubeh Kasiri4
1 Genetics Center, Shariati Hospital, Tehran Medical University, Tehran, Iran
2 Young Researchers Department, Islamic Azad University, Rasht, Iran
3 Sarem Cell Research Centre (SCRC) and Department of Medical Genetics, Sarem Hospital, Tehran, Iran
4 Genetics Salamat Center, Shahr-e Kord, Tehran, Iran
| Date of Web Publication | 30-Oct-2013 |
Correspondence Address:
Sassan Saber
Genetics Center, Shariati Hospital, Tehran Medical University
Iran
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.120813

 Abstract Abstract | | |
Progressive familial intrahepatic cholestasis is an autosomal recessive liver disorder caused by (biallelic) mutations in the ATP8B1 of ABCB11 gene. A nine-year-old girl with cholestasis was referred for genetic counseling. She had a family history of cholestasis in two previous expired siblings. Genetic analysis of the ABCB11 gene led to the identification of a novel homozygous mutation in exon 25. The mutation 3593- A > G lead to a missense mutation at the amino acid level (His1198Arg). This mutation caused PFIC2 due to abnormal function in the bile salt export pump protein (BSEP).
Keywords: ABCB11 gene, novel mutation, progressive familial intrahepatic cholestasis
How to cite this article:
Saber S, Vazifehmand R, Bagherizadeh I, Kasiri M. A novel ABCB11 mutation in an Iranian girl with progressive familial intrahepatic cholestasis. Indian J Hum Genet 2013;19:366-8 |
How to cite this URL:
Saber S, Vazifehmand R, Bagherizadeh I, Kasiri M. A novel ABCB11 mutation in an Iranian girl with progressive familial intrahepatic cholestasis. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:366-8. Available from: http://www.ijhg.com/text.asp?2013/19/3/366/120813 |
| Introduction | |  |
Progressive familial intrahepatic cholestasis (PFIC), known as a group of heterogeneous autosomal recessive disorders, hit liver hepatocellular bile salt secretion as a cholestasis of hepatocellular origin appearing in infants and leading to death from liver failure. Three types of PFIC have been defined from the hepatocellular transport system defects and related genes, which involved in bile formation. [1],[2],[3] Main clinical manifestations include cholestasis, pruritus and jaundice. These patients usually develop fibrosis and end-stage liver disease before adulthood. [4] PFIC type 1 is caused by mutations in ATP8B1 (chromosome 18q21), which encodes FIC1 protein. PFIC type 2 is identified by mutations in ABCB11 (chromosome 2q24), which encodes bile salt export pump (BSEP) and defects in ABCB4-encoding the multi-drug resistant 3 (MDR3) protein - impair biliary phospholipid secretion cause PFIC3. Both PFIC1 and PFIC2 are caused by impaired bile salt secretion appearing in first months of life but PFIC3 is caused by impaired biliary phospholipid secretion and may manifest later. [1],[2]
| Case Report | | |
A nine-year-old girl with cirrhosis was referred for genetic counseling. The patient was affected by icterus and pruritus from newborn period. Her brother and sister displayed the same symptoms; they died at the age of 1.5 and 7 years, respectively. Her parents had a non-consanguinity marriage. The patient's serum biochemical analysis indicated high in total and direct bilirubin and S.G.P.T (ALT). The copper level was raised in serum biochemical result. In CT scan examination, liver and gallbladder had normal sizes but spleen was significantly enlarged. Microscopic examination from a liver biopsy specimen showed significant pathological characterizations of cholestasis. Hepatocytes indicated extensive pseudo acinar transformation; ballooning degeneration, cholestasis and bile plague. Portal tracts contain moderate lymphocytic infiltration and also necrosis in focal confluent. Fibrosis expansion of portal tracts with marked bridging and focal bile duct damage were observed. From the pathological examination of the hepatocytes and clinical demonstrations, the changes were compatible with progressive familial intra-hepatic cholestasis type 1 or 2. The liver biopsy result from the expired sister had the same pathological characteristics of the proband and ended with cirrhosis.
Pathological study
The diagnosis of the kind of PFIC can be difficult because PFIC type 1 has initial clinical and laboratory findings similar to those of PFIC type 2. [1],[2] Pathological features may differ. In PFIC type 1 bland canalicular cholestasis with variable fibrosis is found. In PFIC type 2, variable features include canalicular cholestasis and a neonatal hepatitis pattern, with hepatocellular swelling and giant cell transformation. [1],[2] Immune staining is a useful diagnostic tool for PFIC type 2 since most patients with ABCB11 mutations and hepatobiliary disease of onset in infancy have no canalicular BSEP expression. [5] So pathological study was performed on proband. Hepatocellular swelling with multinucleated canalicular and hepatocellular cholestasis, portal lymphocytic infiltration and fibrosis, newly formed liver nodules, bridging necrosis, bile duct proliferation, giant cells and micro vesicular fat droplet can be seen in proband liver biopsy [Figure 1].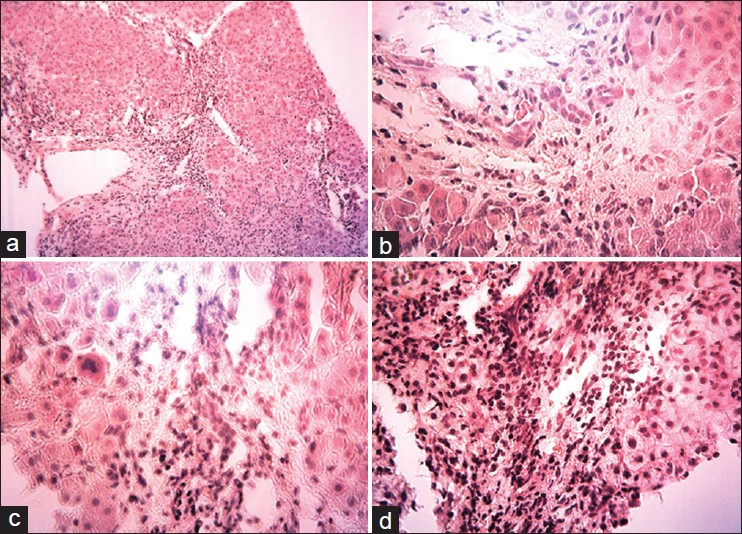 | Figure 1: Liver biopsy, pathologic findings on liver biopsy including (a) Trichrom staining; newly formed liver nodules - Bridging necrosis Porto portal, 10 × 100 (b) Trichrom staining; Bile duct proliferation, 25 × 100 (c) Trichrom staining; Giant cell (arrow head), 40 × 100 (d) Trichrom staining; Hydro pic and microvascular fat droplet, 25 × 100
Click here to view |
| Mutation Analysis | | |
Mutation analysis was performed on DNA of the patient and both parents. The genomic DNA was extracted from peripheral blood leukocytes and ABCB11 of all 27 coding exons and intron/exon borders were screened by intronic oligonucleotide primers as reported previously [6] and direct sequencing of PCR products. A novel homozygous mutation 3593-A > G was detected in exon 25 [Figure 2]. This mutation leads to the missense mutation (His1198Arg). There is a small physicochemical difference between His and Arg, but the amino acid histidine at codon 1198 is highly conserved between species (up till C. elegans) and isoforms (ABCB 1, 4, 5, 6, 7, 8, 9, 11). PolyPhen predicts a probably damaging effect (PSIC score difference: 3.349) and SIFT also predicts that this substitution could affect protein function. Parents were heterozygote for this mutation.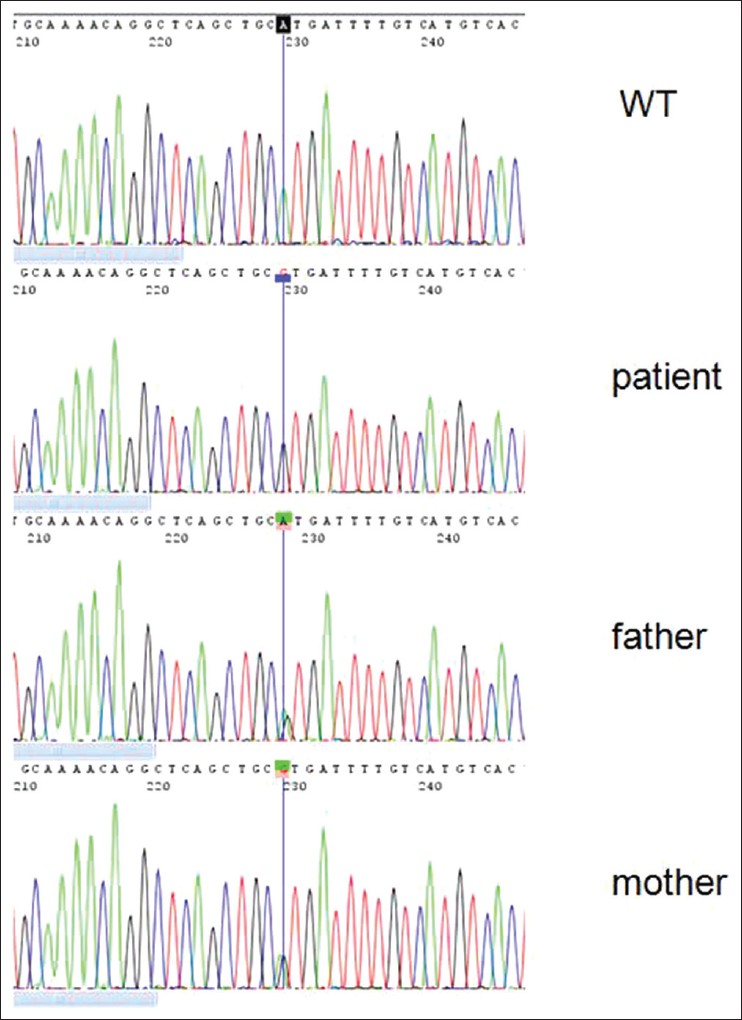 | Figure 2: Sequence chromatograph showing a novel homozygous mutation (3593-A > G) in exon 25 of ABCB11 gene
Click here to view |
| Discussion | | |
To the best of our knowledge, this is the first case report of a patient with typical clinical characterizations of pathological PFIC type 2 in Iran. The patient had the clinical presentation of progressive cholestasis toward the cirrhosis like her two previous siblings expired at the age of 1.5 and 7 years old had affected to the PFIC type 2. The primary diagnosis of the disease was between PFIC type 1 or 2. The most clinical features of the patient and her previous sister are in common. The first clinical characterizations of disease appeared at the early stage of infancy with jaundice incidence. Alanine transferase (ALT) and total and direct bilirubin and serum copper were elevated in serum biochemical result. Liver and gallbladder had normal sizes but spleen size was significantly enlarged. Bile duct proliferation, inflammation, fibrosis or cirrhosis is common features in liver histology of patients with PFIC type 1 and 2. Pathological examination indicated pseudo acinar transformation, ballooning degeneration, cholestasis and bile plague. Portal tracts contain moderate lymphocytic infiltration and also necrosis in focal confluent. Fibrosis expansion of portal tracts with marked bridging and focal bile duct damage were observed. In PFIC type 2, the liver histology shows more inflammatory activity than in PFIC type 1 with giant cell transformation and lobular and portal fibrosis. To date, more than 100 mutation in ABCB11 gene have been identified. [5],[7],[8] Severe phenotypes are often associated with mutations that lead to premature protein truncation or failure of protein production. Insertion, deletion, nonsense, and splicing mutations result in damaging effects, and patients who have clinical PFIC associated with such mutations exhibit little or no BSEP expression in hepatocyte canaliculi. [2],[5] In Asian patients, few reports of mutations in PFIC type 2 exist [9],[10] Goto et al. [8] have reported four mutations in ABCB11 gene, predicated to yield V330X, R487H, R575X and E636G in two Japanese PFIC patients. Chen et al. [10] have reported seven BSEP mutations (M183V, V284L, R303K, R487H, W493X, G1004D and 1145delC) in four PFIC patients of Chinese descent; none of these mutations has been described in Caucasian patients. But here we found a new mutation in ABCB11 gene. This mutation is a novel homozygous mutation that leads to the missense mutation (His1198Arg).
| References | | |
| 1. | Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nut 2008;46:241-52. 
|
| 2. | Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis 2009;4:1.
|
| 3. | Harris MJ, Le Couteur DG, Arias IM. Progressive familial intrahepatic cholestasis: Genetic disorders of biliary transporters. J Gastroenterol Hepatol 2005;20:807-17.
|
| 4. | Whitington PF, Freese DK, Alonso EM, Schwarzenberg SJ, Sharp HL. Clinical and biochemical findings in progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr 1994;18:134-41.
|
| 5. | Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerová D, Rayner A, Dutton L, et al. Servere bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008;134:1203-14.
|
| 6. | Suporn Treepongkaruna, Amornphun Gaensan, Paneeya Pienvichit, Ondrej Luksan, AS Knisely, Pattana Sornmayura, and Milan Jirsa. Novel ABCB11 mutations in a Thai infant with progressive familial intrahepatic cholestasis, World J Gastroenterol. 2009 September 14;15(34):4339-4342.
|
| 7. | Pauli-Magnus C, Stieger B, Meier Y, Kullak-Ublick GA, Meier PJ. Enterohepatic transport of bile salts and genetics of cholestasis. J Hepatol 2005;43:342-57.
|
| 8. | Goto K, Sugiyama K, Sugiura T, Ando T, Mizutani F, Terabe K, et al. Bile salt export pump gene mutations in two Japanese patients with progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr 2003;36:647-50.
|
| 9. | Chen HL, Chang PS, Hsu HC, Ni YH, Hsu HY, Lee JH, et al. FIC1 and BSEP defects in Taiwanese patients with chronic intrahepatic cholestasis with low gamma-glutamyltranspeptidase levels. J Pediatr 2002;140:119-24.
|
| 10. | Chen HL, Liu YJ, Su YN, Wang NY, Wu SH, Ni YH, et al. Diagnosis of BSEP/ABCB11 mutations in Asian patients with cholestasis using denaturing high performance liquid chromatography. J Pediatr 2008;153:825-32.
|
[Figure 1], [Figure 2]
|









