|
 
 |
|
REVIEW ARTICLE |
|
|
|
| Year : 2014 | Volume
: 20
| Issue : 1 | Page : 10-19 |
| |
Reactive metabolites and antioxidant gene polymorphisms in type 2 diabetes mellitus
Monisha Banerjee, Pushpank Vats
Department of Zoology, Molecular and Human Genetics Laboratory, University of Lucknow, Lucknow, Uttar Pradesh, India
| Date of Web Publication | 19-May-2014 |
Correspondence Address:
Monisha Banerjee
Department of Zoology, Molecular and Human Genetics Laboratory, University of Lucknow, Lucknow 226 007, Uttar Pradesh
India
 Source of Support: None, Conflict of Interest: None  | 8 |
DOI: 10.4103/0971-6866.132747

 Abstract Abstract | | |
Type 2 diabetes mellitus (T2DM), by definition is a heterogeneous, multifactorial, polygenic syndrome which results from insulin receptor (IR) dysfunction. It is an outcome of oxidative stress caused by interactions of reactive metabolites (RMs) with lipids, proteins and other molecules of the human body. Production of RMs mainly superoxides (•O2− ) has been found in a variety of predominating cellular enzyme systems including nicotinamide adenine dinucleotide phosphate oxidase, xanthine oxidase, cyclooxygenase, endothelial nitric oxide synthase (eNOS) and myeloperoxidase. The four main RM related molecular mechanisms are: increased polyol pathway flux; increased advanced glycation end-product formation; activation of protein kinase C isoforms and increased hexosamine pathway flux which have been implicated in glucose-mediated vascular damage. Superoxide dismutase, catalase, glutathione peroxidase, glutathione-S-transferase and NOS are antioxidant enzymes involved in scavenging RMs in normal individuals. Functional polymorphisms of these antioxidant enzymes have been reported to be involved in the pathogenesis of T2DM. The low levels of antioxidant enzymes or their non-functionality results in excessive RMs which initiates stress related pathways thereby leading to IR and T2DM. An attempt has been made to review the role of RMs and antioxidant enzymes in oxidative stress resulting in T2DM.
Keywords: Antioxidants, oxidative stress, polymorphisms, reactive metabolites, type 2 diabetes mellitus
How to cite this article:
Banerjee M, Vats P. Reactive metabolites and antioxidant gene polymorphisms in type 2 diabetes mellitus. Indian J Hum Genet 2014;20:10-9 |
How to cite this URL:
Banerjee M, Vats P. Reactive metabolites and antioxidant gene polymorphisms in type 2 diabetes mellitus. Indian J Hum Genet [serial online] 2014 [cited 2016 Aug 24];20:10-9. Available from: http://www.ijhg.com/text.asp?2014/20/1/10/132747 |
| Introduction | |  |
Diabetes mellitus (DM) is a chronic disorder characterized by impaired metabolism of glucose and lipids due to defect in insulin secretion (beta cell dysfunction) or action (insulin resistance [IR]). The characteristic properties of DM are hyperglycemia, microvascular (e.g., retina, renal glomerulus and peripheral nerve) as well as macrovascular (e.g., atherosclerosis, coronary artery disease [CAD], stroke) pathologies with more than 17.5 million deaths world-wide. [1] Oxidative stress has been implicated as the underlying cause of both macrovascular and microvascular complications associated with type 2 diabetes mellitus (T2DM). It is believed that therapies aimed at reducing oxidative stress would benefit patients with T2DM and also those at risk. The accumulation of glucose and fatty acids within muscles adipose tissue and pancreatic cells combined with a sedentary life-style lead to the generation of excess reactive metabolites (RMs). Oxidative stress and RMs are interrelated terms defined in general as excess formation and/or insufficient removal of highly reactive molecules such as reactive oxygen species (ROS), reactive nitrogen species (RNS) and reactive thiyl species (reactive thiyl and tyrosyl radicals
[RTR]). According to International Diabetes Federation Diabetes Atlas 5 th Edition-2012 update, 371 million people have been reported with DM and the number is expected to rise to > 552 million by 2030. The 2012 Indian statistics showed 63.0 million diabetic cases and prevalence of 8.37% in the adult population . [2] Moreover, a 4.0% prevalence of T2DM was reported in North Indian population. [3] This short review aims to explain the role of oxidative stress leading to IR, β-cell dysfunction, impaired glucose tolerance and ultimately T2DM and also the association of antioxidant gene polymorphisms.
| Types of RMs | | |
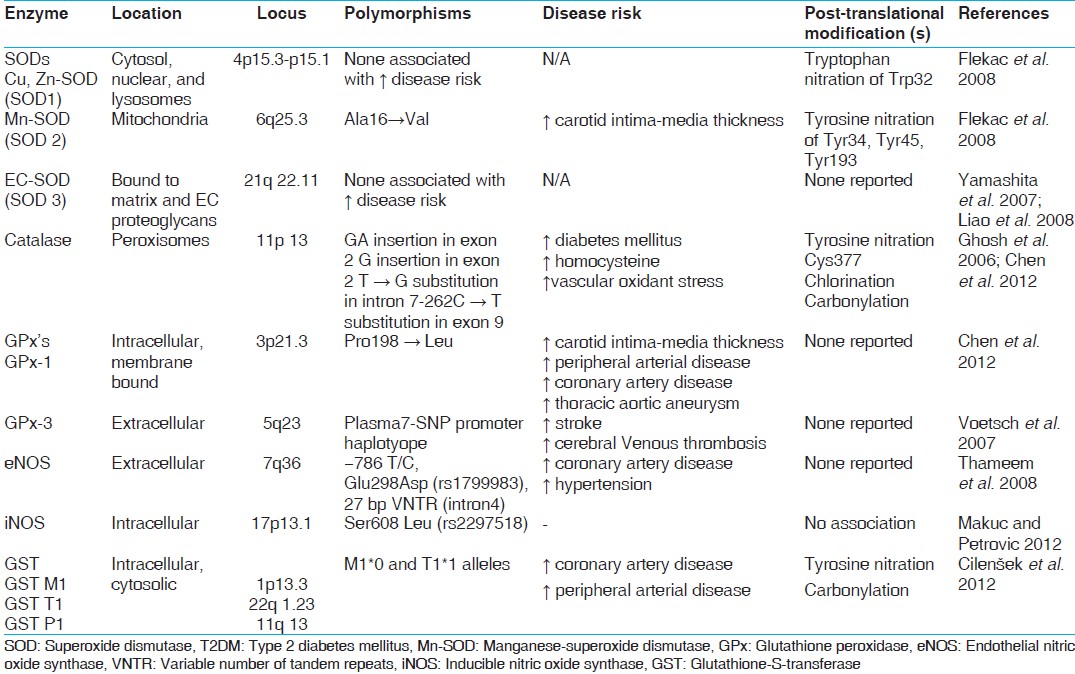
[Table 1]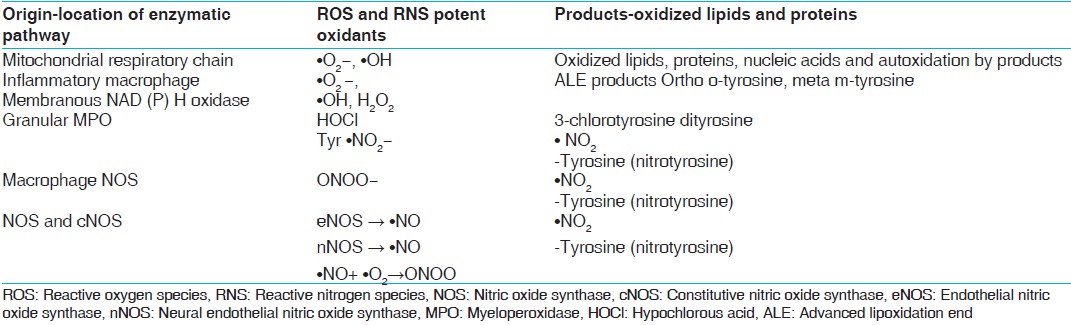 | Table 1: Enzymatic pathways: Origin, ROS, RNS and their products[48,49]
Click here to view |
ROS
Oxygen derived free radicals (ODFR) and oxygen derived non-radicals (ODNR) are generated in metabolic pathways of biological systems. ODFR include superoxide (•O 2− ), hydroxyl (•OH), peroxyl (•RO 2 ), hydroperoxyl (•HRO 2− ) while ODNR include hydrogen peroxide (H 2 O 2 ) and hydrochlorous acid (HOCl). These metabolites are responsible for lipid and protein modifications in case of oxidative stress. Basal oxidative cellular metabolism generates a number of ODFR species through the activation of enzymes that produce superoxide anions and/or byproducts of mitochondrial respiration. [4]
RNS
Like ROS, RNS can be classified into radical and non-radical species, nitrogen derived free radicals (NDFR) and nitrogen derived non-radicals (NDNR). NDFR include nitric oxide (•NO), nitrogen dioxide (•NO 2− ) while NDNR include alkyl peroxynitrates (RONOO− ), nitrous oxide (HNO 2 ) and peroxynitrite (ONOO− ). •O 2− •NO and ONOO− are the most widely studied species and play important roles in cardiovascular complications. [4] Nitric oxide (•NO) is responsible for the formation of many end-products involved in oxidative stress directly or indirectly after reaction with oxygen. •NO-derived RNS react with aromatic amino acids, lipids and thiols resulting in lipid and protein modifications. Leukocyte peroxidases are involved in the formation of •NO 2 after utilization of H 2 O 2 and NO 2− as substrates. •NO 2 , •OH and ONOOH are responsible for damages related to oxidative stress e.g., oxidation, nitrosation and nitration reactions. [4],[5]
RTR
Thiyl radicals (TR) may be formed by •OH, ONOO− and/or Fe 3+ mediated oxidation of thiols. TR may also be derived from sulfur containing moieties including disulfide, thioester or thioether functionalities under conditions of oxidative stress. Once formed, TR not only reacts with themselves and oxygen but also oxidize biological electron donors including ascorbic acid, nicotinamide adenine dinucleotide and ferricytochrome C.
| Production of RMs | | |
Production of RMs mainly superoxides (•O 2− ) have been found in a variety of predominating cellular enzyme systems including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase (XO), cyclooxygenase (COX), uncoupled endothelial nitric oxide synthase (eNOS) and myeloperoxidase (MPO) [Table 1]. [6] The various sources of ROS and action of antioxidant enzymes have been represented in [Figure 1]. NADPH oxidase uses NADPH as a substrate and is considered as an important source of ROS generation in vascular cells. The lipoxygenase (LPO) and COX generate ROS indirectly by promoting the formation of inflammatory mediators. RM production may result from action of arachidonic acid (AA) metabolizing enzymes including cytochrome P-450, LPO, COX and those in the mitochondrial respiratory chain. [7] AA is cleaved from the membrane by phospholipase A2 and is then metabolized by 5-LPO in the presence of its accessory protein 5-LPO activating protein to form leukotrienes. AA is also metabolized by COX to form members of another family of inflammatory mediators, the prostaglandins. Mitochondria also generate superoxides as electrons are transferred from complexes I to IV during normal cellular respiration. XO, which converts hypoxanthine and xanthine to uric acid, is an additional source of ROS. Finally, eNOS uncouples to generate superoxide in preference to NO. [8]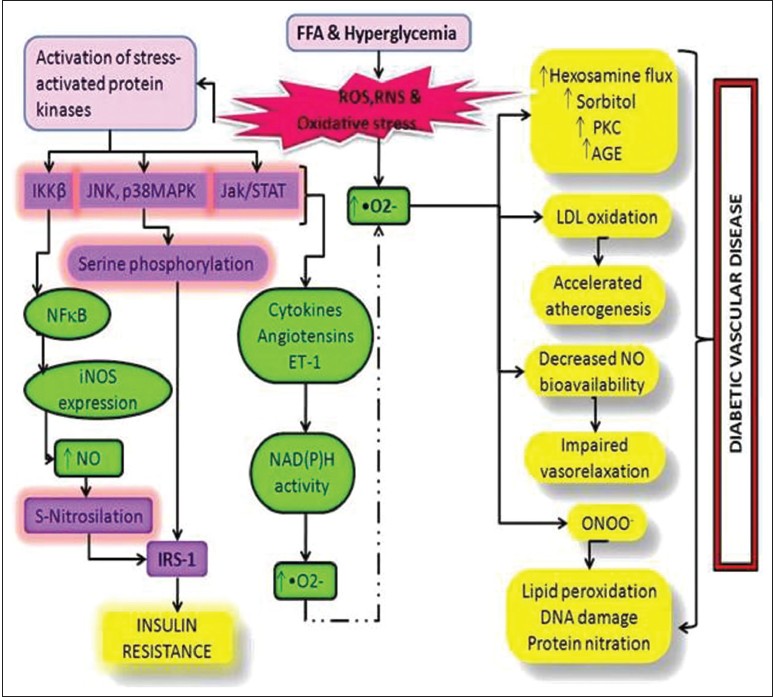 | Figure 1: Schematic representation of oxidative stress and the pathways leading to type 2 diabetes mellitus and its complications
Click here to view |
| RMs and T2DM | | |
ROS, •O 2− leads to several damaging pathways resulting in micro and macrovascular complications in diabetes. There are four main molecular mechanisms implicated in glucose-mediated vascular damage viz., increased polyol pathway flux; increased production of advanced glycation end-product; activation of protein kinase C (PKC) isoforms, sorbitol, cytokines and prostanoids along with increased hexosamine pathway flux [Figure 2]. •O 2− and H 2 O 2 stimulate stress-related signaling mechanisms such as nuclear factor kappa light chain enhancer of activated B cells (NF-κB), p38 mitogen activated protein kinase and Janus kinase signal transducer and activator of transcription resulting in vascular smooth muscle cell (VSMC) migration and proliferation. H 2 O 2 also mediates apoptosis and pathological angiogenesis in endothelial cells. [5] Pathway-selective IR also results in decreased endothelial production of the anti-atherogenic molecule, nitric oxide. Additional stress-sensitive kinases that are reported to be involved in IR substrate (IRS)-mediated IR include several isozymes of PKC such as PKCβ, PKCγ and inhibitor kinase beta NF-κB. [9] Once activated, these kinases are able to phosphorylate multiple targets including IR and IRS proteins such as IRS-1 and IRS-2. Oxidative stress on IRS serine phosphorylation can lead to IR. [10] This reflects decreased activity of vasodilators such as nitric oxide, increased activity of vasoconstrictors such as angiotensin II and endothelin-1 and elaboration of permeability factors such as vascular endothelial growth factor. Quantitative and qualitative abnormalities of extracellular (EC) matrix contribute to an irreversible increase in vascular permeability. Microvascular cell loss occurs with time as a result of programmed cell death and progressive capillary occlusion. Both occur due to extracellular matrix and overproduction induced by growth factors such as transforming growth factor-β and deposition of plasma proteins. [8] A causative link among hyperglycemia, mitochondrial ROS generation, oxidative stress and development of complications has been suggested which plays a key role in the pathogenesis of diabetes. Damage due to RMs is associated with complex metabolic and structural changes in the body for example, oxidation of low-density lipoproteins (Ox-LDL) which are taken up by scavenger receptors in macrophages leading to foam cell formation and atherosclerotic plaques. [11],[12] ROS-induced peroxidation of membrane lipids alters the structure and fluidity of biological membranes which ultimately affects cellular function. [13]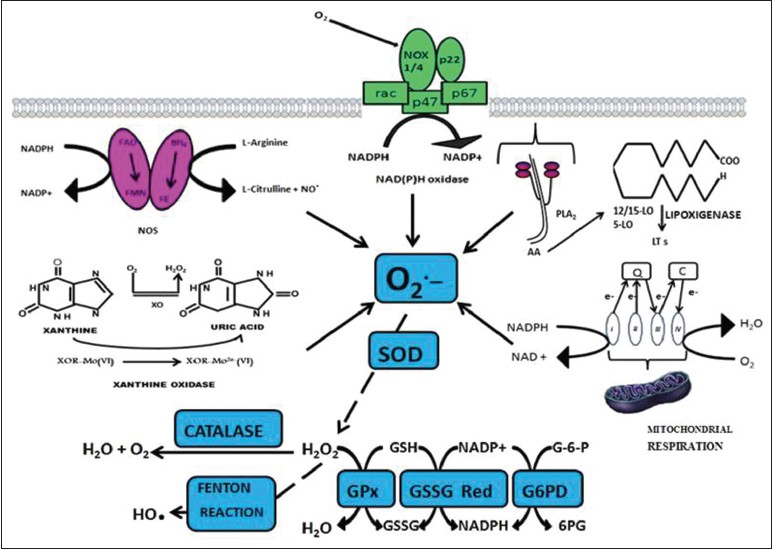 | Figure 2: Outline of various sources of reactive oxygen species and action of antioxidant enzymes. Q: Indicates coenzyme Q; C: Cytochrome C; FAD: Flavin adenine dinucleotide; FMN: Flavin mononucleotide; FE: Heme iron; BH4: Tetrahydrobiopterin
Click here to view |
In T2DM, such activation of stress-sensitive pathways and elevation in glucose and free fatty acid levels lead both to IR and impaired insulin secretion. Early in the course of diabetes, intracellular hyperglycemia causes abnormalities in blood flow and increases vascular permeability. In diabetic arteries, endothelial dysfunction seems to involve both IR specific to the phosphatidylinositol-3-OH kinase pathway and hyperglycemia. Both IR and hyperglycemia have also been implicated in the pathogenesis of diabetic dyslipidemia. Hyperglycemia seems to cause raised levels of atherogenic cholesterol-enriched apolipoprotein B-containing remnant particles by reducing expression of heparan sulphate proteoglycan and perlecan on hepatocytes. Hyperglycemia may also decrease production of trophic factors for endothelial and neuronal cells. Together, these changes lead to edema, ischemia and hypoxia-induced neovascularization in the retina, proteinuria, mesanglial matrix expansion and glomerulosclerosis in the kidney and multifocal axonal degeneration in peripheral nerves. Therefore, as a consequence of its microvascular pathology, diabetes is the leading cause of blindness, end stage renal disease and a variety of debilitating neuropathies. The pathogenesis of atherosclerosis also begins with endothelial dysfunction. Postprandial hyperglycemia may be more predictive of atherosclerosis than fasting plasma glucose level or hemoglobin A1c. Atherosclerotic macrovascular disease affects arteries that supply the heart, brain and lower extremities. As a result, patients with diabetes have a much higher risk of myocardial infarction, stroke and limb amputation. [14]
In healthy individuals both enzymatic and non-enzymatic antioxidant defense play important roles in scavenging ROS and RNS [Table 2]. Impaired antioxidant defense increases oxidative stress and contributes to the development of T2DM and diabetic cardiovascular disease (CVD). •O 2− produces H 2 O 2 on its dismutation by copper superoxide dismutase (Cu-SOD) and manganese-SOD (Mn-SOD). H 2 O 2 produces hydroxyl radical (•OH) by reaction with reduced transition metals (Fe or Cu) i.e. fenton reaction and can be metabolized to HOCl by MPO. [6] H 2 O 2 is converted to H2 O and O2 by glutathione peroxidase (GSH-Px) or catalase (CAT) in the mitochondria and lysosomes respectively. The inner mitochondrial membrane also contains vitamin E which is a powerful antioxidant as it can accept unpaired electrons to produce a stable product. [14]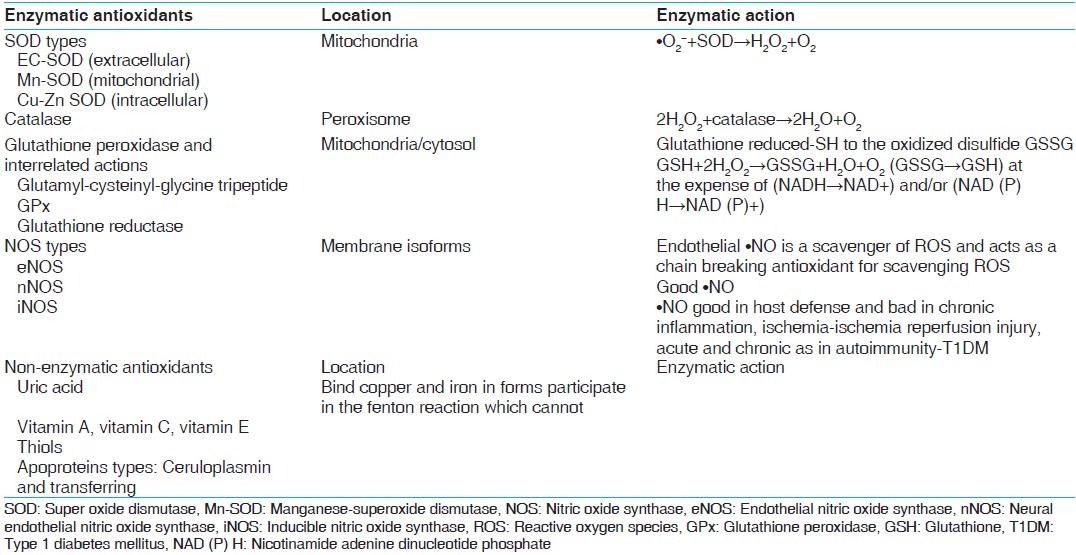 | Table 2: Antioxidants in catalytic/enzymatic inactivation of free radicals and non - enzymatic antioxidants[48,49]
Click here to view |
| Antioxidant Gene Polymorphism | | |
[Table 2] and [Table 3]
SOD
As mentioned earlier, once formed the •O2− is dismutated enzymatically to H2 O2 and oxygen by the SOD family of antioxidant enzymes which include intracellular (Cu Zn-SOD), mitochondrial (Mn-SOD) and extracellular (EC-SOD) enzymes also referred to as SOD types 1, 2 and 3 respectively. [8] An increase in Cu Zn-SOD (SOD1) expression projects human smooth muscle cells against oxidative injury. Ox-LDL causes an increase in the DNA binding activity of activator protein-1 and NF-κB which is inhibited by Cu Zn-SOD overexpression. Cu Zn-SOD (SOD1) gene is located on chromosome 4p15.1-15.3 while Mn-SOD (SOD2) gene is located on 6q25. A functional polymorphism in exon 2 of SOD2 gene A16V (C/T) (rs4880) was identified that resulted in structural alterations in the mitochondrial targeting domain, implicating its decreased antioxidant potential to limited post-transcriptional transport. The substitution from valine to alanine was shown to induce a 30-40% increase in Mn-SOD activity in mitochondria.
Individuals harboring this variant had an increased carotid intima-to-media thickness (IMT) and were at increased risk for CAD and acute myocardial infarction. [15] Increased vascular oxidative stress also led to decreased SOD activity through post-translational modification of the enzyme and Mn-SOD polymorphism affected Ox-LDL induced apoptosis and CAD. [16] Polymorphic conditions of SOD1 35 A/C (rs2234694) and SOD2 A16V (C/T) showed that serum SOD activity was higher in individuals with CC genotype than TT genotype of SOD2 gene and higher in AA when compared to CC genotype of SOD1 gene. Better diabetes control was found in patients with CC genotype of SOD2 gene. Significantly different allele and genotype frequencies of SOD2 gene polymorphism were found among T2DM patients with and without macroangiopathy, diabetic retinopathy (DR) in Chinese, diabetic macular edema and albuminuria in Koreans. [17],[18],[19]
EC-SOD (SOD3) is bound to matrix and EC proteoglycans. EC-SOD (SOD3) gene is located on chromosome 21q22.11. Clinical studies have shown a decrease in EC-SOD activity in aged, African-Americans with hypertension, patients with vasospastic angina, thoracic aortic aneurysm and calcified aortic stenosis. [20],[21] Serum SOD activity was significantly decreased in T2DM subjects compared to control subjects. The variant R213G in the heparin-binding domain of EC-SOD has been linked to CVD risk. This polymorphism was linked to increased risk of ischemic heart disease (IHD) in a Danish case control study as well. [22]
CAT
CAT is present in peroxisomes and exists as a dumbbell-shaped tetramer of four identical subunits. It rapidly catalyzes the decomposition of H2 O2 into less reactive oxygen and water molecules. Deficiency of this enzyme was known to lead to T2DM development. [23] Exon 2 and neighboring introns of the CAT gene located on chromosome 11p13 were thought to be mutation hot spots for T2DM susceptibility. [24],[25] Under conditions of oxidative stress, modification of cystein to cysteic acid leads to tyrosyl nitration of CAT and its decreased activity. [26],[27] The − 262C/T promoter polymorphism in CAT gene was examined in types 1, 2 and gestational diabetes and complications such as DR, diabetic nephropathy (DN), IHD and CVD. [28],[29] The C111T functional polymorphism in exon 9 of CAT gene contributed to the CAT activity in different types of T2DM. [29],[30]
GPx
GPx are selenocysteine-containing enzymes that catalyze the reduction of H2 O2, lipid hydroperoxides to H2 O and lipid alcohols respectively in a reaction that utilizes GSH as a reducing co-substrate. [28] There are 5 known forms of GPx: cellular (GPx-1), gastrointestinal (GPx-2), plasma (GPx-3), phospholipid (GPx-4) and sperm (snGPx). The importance of GPx family of antioxidant enzymes limits the oxidative risk for atherothrombosis. GPx-1 is an ubiquitous antioxidant enzyme whose deficiency has been shown to promote endothelial dysfunction, heart failure and abnormal structural changes in vasculature and myocardium. [31] Interestingly, hyperhomocystinemia appears to enhance vascular oxidative stress and atherothrombosis, in part by suppressing expression of the GPx-1 gene which is located on chromosome 3p21.3. Erythrocyte GPx-1 activity and association of GPx-1 genotypes was shown as independent determinants of cardiovascular risk and CAD. [32] A polyalanine sequence polymorphism in exon 1 of GPx-1 gene produces 3 alleles with 5, 6, or 7 alanine repeats. Men with at least one 6-alanine repeat had significantly increased risk of CAD.
The Pro198 Leu C/T polymorphism in GPx-1 gene increased carotid IMT, prevalence of cardiovascular and peripheral vascular disease in Japanese patients with T2DM. [28],[33] Out of a large number of factors mediating atherosclerotic risk in plasma, focus lays on GPx-3, the essential extracellular peroxidase and its role in modulating oxidative stress. Deficiency of GPx-3 was associated with decreased nitric oxide bioavailability and increased platelet dependent thrombosis. There was a reduction in plasma GPx-3 activity with increased platelet activation and cerebrovascular arterial thrombosis. [34],[35] GPx-3 promoter revealed seven polymorphisms that are tightly linked and form two novel haplotypes, out of which one was associated with hypoxic conditions, arterial thrombotic stroke and cerebral venous thrombosis. [36] Over expression of GPx-4 reduces oxidized phospholipids, cholesterol hydroperoxides as well as proinflammatory lipid peroxides generated by LPO and COX thereby decreasing vascular oxidative stress and progression of atherosclerosis. [37]
Glutathione-S-transferases (GST)
The GSTs are dimeric cytosolic xenobiotic-metabolizing enzymes that catalyze the conjugation of an active xenobiotic to GSH, an endogenous water-soluble substrate and detoxify reactive electrophiles such as those contained in tobacco smoke. In addition to their catalytic role in detoxification, GSTs were also found to possess selenium-independent peroxidase activity with hydroperoxides, steroid isomerization capacity, binding and transport of bilirubin, heme, bile salts and steroids in a process that is associated with a decrease in enzymatic activity. [38] Several studies have found an association between GST polymorphisms and decreased enzymatic activity and atherosclerosis. Elevated levels of plaque DNA damage as well as levels of inflammatory markers such as C-reactive protein, fibrinogen and adhesion molecules were detected in individuals with GSTM1*0 null allele.
GST M1, T1 and P1 have been reported to be involved in T2DM development and various diabetes related complications. [39],[40],[41],[42],[43] Microsomal GST3 encoded by MGST3 gene, which maps to chromosome 1q23 is a potential susceptibility gene linked to T2DM in Pima Indians, Caucasian and Chinese populations. [44]
NOS
Nitric oxide (NO) plays a fundamental role in the regulation of endothelial function and vascular tone in many organs including kidney. It inhibits platelet aggregation, leukocyte adhesion to vascular endothelium and Ox-LDL. [45] Upon release, NO diffuses rapidly through the cell membrane and relaxes neighboring vascular smooth cells through the production of cyclic guanine 3'5'- monophosphate (cGMP). cGMP then activates the protein kinase G family, leading to a cascade of responses at the levels of transcription and translation. Inducible nitric oxide synthase (NOSI/iNOS), neural NOS (NOS II/nNOS) and NOSIII/eNOS are the three isoforms of NOS. Clinically, eNOS uncoupling has been associated with hypertension, DM, hypercholesterolemia and atherosclerosis. [46] Impairment of NO production causes endothelial dysfunction which contributes to the development of IR, T2DM, chronic renal failure and cardiovascular complications including hypertension and hypercholesterolemia. [47]
eNOS or NOSIII gene, mapped to chromosome 7q36 is highly polymorphic and several studies have been undertaken to investigate the potential association of polymorphisms and risk of atherothrombotic vascular disease in Caucasian and Asian populations. SNPs in the promoter region (−786 T/C), G/T substitution at nucleotide 894 in exon 7 leading to an amino acid change (Glu298Asp) and a 27 bp variable number of tandem repeats in intron 4 have received much attention because of their functional relevance to eNOS activity and association with cardiovascular and renal diseases. [45] The Glu298Asp gene polymorphism is responsible for a decrease in basal NO production and increased frequency of hypertension. [48] Additionally, eNOS Glu298Asp can interact with gene polymorphisms of other endogenous antioxidant enzymes in T2DM patients. [48],[49],[50],[51]
In atherosclerosis, VSMC, monocytes, macrophages and dendritic cells all express iNOS. Induction of iNOS may occur following exposure to inflammatory cytokines, including interleukin-1 β, interferon-γ and tumor necrosis factor-α. In contrast to eNOS, iNOS binds Ca 2+ /calmodulin tightly and does not require an increase in intracellular Ca 2+ for activation. [52] The presence of iNOS localized to macrophages and VSMC was found to colocalize with oxidized lipid and protein derivatives found in atherosclerotic plaques. [53] However, no association of eNOS Glu298Asp (rs1799983), eNOS 4a/b and iNOS Ser608 Leu (rs2297518) polymorphisms was found in T2DM patients with DN. [54]
| Conclusions | | |
T2DM and oxidative stress have both clinical and genetic correlation required to be established to a greater extent. The overall play of the RMs has shown to be a leading cause of late onset IR. These RMs are generated inside the human body in a scheduled manner in normal individuals and have a feedback control with antioxidant system. Glycemic load and RMs are highly interrelated leading to various harmful molecular mechanisms implicated in glucose-mediated vascular damage. The various pathways lead to increased expression of other signals which result in IR and T2DM. This short review undertaken to understand the role of RMs in T2DM and IR will provide a lead for future research in identifying genes of oxidative stress control pathways. Furthermore, the proper understanding of antioxidant gene polymorphisms and their association with T2DM may lead to the development of prognostic markers in the near future.
| Acknowledgments | | |
The authors are grateful to the funding agencies viz. Indian Council of Medical Research (ICMR) and Department of Science and Technology, New Delhi, India for generous grants. PV is thankful to ICMR for Junior Research Fellowship.
| References | | |
| 1. | Moore DJ, Gregory JM, Kumah-Crystal YA, Simmons JH. Mitigating micro- and macro-vascular complications of diabetes beginning in adolescence. Vasc Health Risk Manag 2009;5:1015-31. 
|
| 2. | International Diabetes Federation (IDF). Diabetes Atlas. 5 th ed. USA: World Diabetes Foundation; 2012.
|
| 3. | Banerjee M, Saxena M. An overview and molecular genetics of type 2 diabetes mellitus. In: Type 2 Diabetes Mellitus: Causes, Treatment and Preventive Strategies. New York: Nova Science Publishers Inc.; 2012. p. 1-64.
|
| 4. | Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: Linking basic science to clinical practice. Cardiovasc Diabetol 2005;4:5.
|
| 5. | Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension 2003;42:1075-81.
|
| 6. | Woods AA, Linton SM, Davies MJ. Detection of HOCl-mediated protein oxidation products in the extracellular matrix of human atherosclerotic plaques. Biochem J 2003;370:729-35.
|
| 7. | Mueller CF, Laude K, McNally JS, Harrison DG. ATVB in focus: Redox mechanisms in blood vessels. Arterioscler Thromb Vasc Biol 2005;25:274-8.
|
| 8. | Leopold JA, Loscalzo J. Oxidative risk for atherothrombotic cardiovascular disease. Free Radic Biol Med 2009;47:1673-706.
|
| 9. | Dey D, Mukherjee M, Basu D, Datta M, Roy SS, Bandyopadhyay A, et al. Inhibition of insulin receptor gene expression and insulin signaling by fatty acid: Interplay of PKC isoforms therein. Cell Physiol Biochem 2005;16:217-28.
|
| 10. | Bloch-Damti A, Potashnik R, Gual P, Le Marchand-Brustel Y, Tanti JF, Rudich A, et al. Differential effects of IRS1 phosphorylated on Ser307 or Ser632 in the induction of insulin resistance by oxidative stress. Diabetologia 2006;49:2463-73.
|
| 11. | Antonov AS, Kolodgie FD, Munn DH, Gerrity RG. Regulation of macrophage foam cell formation by alphaVbeta3 integrin: Potential role in human atherosclerosis. Am J Pathol 2004;165:247-58.
|
| 12. | Gautam S, Banerjee M. The macrophage Ox-LDL receptor, CD36 and its association with type II diabetes mellitus. Mol Genet Metab 2011;102:389-98.
|
| 13. | Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part I: Basic mechanisms and in vivo monitoring of ROS. Circulation 2003;108:1912-6.
|
| 14. | Newsholme P, Haber EP, Hirabara SM, Rebelato EL, Procopio J, Morgan D, et al. Diabetes associated cell stress and dysfunction: Role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol 2007;583:9-24.
|
| 15. | Kakko S, Päivänsalo M, Koistinen P, Kesäniemi YA, Kinnula VL, Savolainen MJ. The signal sequence polymorphism of the MnSOD gene is associated with the degree of carotid atherosclerosis. Atherosclerosis 2003;168:147-52.
|
| 16. | Fujimoto H, Taguchi J, Imai Y, Ayabe S, Hashimoto H, Kobayashi H, et al. Manganese superoxide dismutase polymorphism affects the oxidized low-density lipoprotein-induced apoptosis of macrophages and coronary artery disease. Eur Heart J 2008;29:1267-74.
|
| 17. | Flekac M, Skrha J, Hilgertova J, Lacinova Z, Jarolimkova M. Gene polymorphisms of superoxide dismutases and catalase in diabetes mellitus. BMC Med Genet 2008;9:30.
|
| 18. | Pácal L, Varvaøovská J, Rušavý Z, Lacigová S, Stìtina R, Racek J, et al . Parameters of oxidative stress, DNA damage and DNA repair in type 1 and type 2 diabetes mellitus. Arch Physiol Biochem 2011;117:222-30.
|
| 19. | Lee SJ, Choi MG. Association of manganese superoxide dismutase gene polymorphism (V16A) with diabetic macular edema in Korean type 2 diabetic patients. Metabolism 2006;55:1681-8.
|
| 20. | Yamashita K, Takahiro K, Kamezaki F, Adachi T, Tasaki H. Decreased plasma extracellular superoxide dismutase level in patients with vasospastic angina. Atherosclerosis 2007;191:147-52.
|
| 21. | Liao M, Liu Z, Bao J, Zhao Z, Hu J, Feng X, et al. A proteomic study of the aortic media in human thoracic aortic dissection: Implication for oxidative stress. J Thorac Cardiovasc Surg 2008;136:65-72, 72.e1-3.
|
| 22. | Juul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R, Sillesen H, et al. Genetically reduced antioxidative protection and increased ischemic heart disease risk: The Copenhagen City Heart Study. Circulation 2004;109:59-65.
|
| 23. | Góth L, Lenkey A, Bigler WN. Blood catalase deficiency and diabetes in Hungary. Diabetes Care 2001;24:1839-40.
|
| 24. | Forsberg L, Lyrenäs L, de Faire U, Morgenstern R. A common functional C-T substitution polymorphism in the promoter region of the human catalase gene influences transcription factor binding, reporter gene transcription and is correlated to blood catalase levels. Free Radic Biol Med 2001;30:500-5.
|
| 25. | Vitai M, Fátrai S, Rass P, Csordás M, Tarnai I. Simple PCR heteroduplex, SSCP mutation screening methods for the detection of novel catalase mutations in Hungarian patients with type 2 diabetes mellitus. Clin Chem Lab Med 2005;43:1346-50.
|
| 26. | Hirono A, Sasaya-Hamada F, Kanno H, Fujii H, Yoshida T, Miwa S. A novel human catalase mutation (358 T - >del) causing Japanese-type acatalasemia. Blood Cells Mol Dis 1995;21:232-4.
|
| 27. | Ghosh S, Janocha AJ, Aronica MA, Swaidani S, Comhair SA, Xu W, et al. Nitrotyrosine proteome survey in asthma identifies oxidative mechanism of catalase inactivation. J Immunol 2006;176:5587-97.
|
| 28. | Chen H, Yu M, Li M, Zhao R, Zhu Q, Zhou W, et al. Polymorphic variations in manganese superoxide dismutase (MnSOD), glutathione peroxidase-1 (GPX1), and catalase (CAT) contribute to elevated plasma triglyceride levels in Chinese patients with type 2 diabetes or diabetic cardiovascular disease. Mol Cell Biochem 2012;363:85-91.
|
| 29. | dos Santos KG, Canani LH, Gross JL, Tschiedel B, Souto KE, Roisenberg I. The catalase -262C/T promoter polymorphism and diabetic complications in Caucasians with type 2 diabetes. Dis Markers 2006;22:355-9.
|
| 30. | Tarnai I, Csordás M, Sükei E, Shemirani AH, Káplár M, Góth L. Effect of C111T polymorphism in exon 9 of the catalase gene on blood catalase activity in different types of diabetes mellitus. Free Radic Res 2007;41:806-11.
|
| 31. | Prabhakar R, Morokuma K, Musaev DG. Peroxynitrite reductase activity of selenoprotein glutathione peroxidase: A computational study. Biochemistry 2006;45:6967-77.
|
| 32. | Blankenberg S, Rupprecht HJ, Bickel C, Torzewski M, Hafner G, Tiret L, et al. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N Engl J Med 2003;349:1605-13.
|
| 33. | Winter JP, Gong Y, Grant PJ, Wild CP. Glutathione peroxidase 1 genotype is associated with an increased risk of coronary artery disease. Coron Artery Dis 2003;14:149-53.
|
| 34. | Ramprasath T, Senthil Murugan P, Prabakaran AD, Gomathi P, Rathinavel A, Selvam GS. Potential risk modifications of GSTT1, GSTM1 and GSTP1 (glutathione-S-transferases) variants and their association to CAD in patients with type-2 diabetes. Biochem Biophys Res Commun 2011;407:49-53.
|
| 35. | Ramprasath T, Murugan PS, Kalaiarasan E, Gomathi P, Rathinavel A, Selvam GS. Genetic association of Glutathione peroxidase-1 (GPx-1) and NAD (P) H: Quinone Oxidoreductase 1(NQO1) variants and their association of CAD in patients with type-2 diabetes. Mol Cell Biochem 2012;361:143-50.
|
| 36. | Voetsch B, Jin RC, Bierl C, Benke KS, Kenet G, Simioni P, et al. Promoter polymorphisms in the plasma glutathione peroxidase (GPx-3) gene: A novel risk factor for arterial ischemic stroke among young adults and children. Stroke 2007;38:41-9.
|
| 37. | Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 2008;8:237-48.
|
| 38. | Loscalzo J. Membrane redox state and apoptosis: Death by peroxide. Cell Metab 2008;8:182-3.
[PUBMED] |
| 39. | Miller EA, Pankow JS, Millikan RC, Bray MS, Ballantyne CM, Bell DA, et al. Glutathione-S-transferase genotypes, smoking, and their association with markers of inflammation, hemostasis, and endothelial function: The atherosclerosis risk in communities (ARIC) study. Atherosclerosis 2003;171:265-72.
|
| 40. | Oniki K, Umemoto Y, Nagata R, Hori M, Mihara S, Marubayashi T, et al. Glutathione S-transferase A1 polymorphism as a risk factor for smoking-related type 2 diabetes among Japanese. Toxicol Lett 2008;178:143-5.
|
| 41. | Bid HK, Konwar R, Saxena M, Chaudhari P, Agrawal CG, Banerjee M. Association of glutathione S-transferase (GSTM1, T1 and P1) gene polymorphisms with type 2 diabetes mellitus in north Indian population. J Postgrad Med 2010;56:176-81.
[PUBMED]  |
| 42. | Amer MA, Ghattas MH, Abo-Elmatty DM, Abou-El-Ela SH. Influence of glutathione S-transferase polymorphisms on type-2 diabetes mellitus risk. Genet Mol Res 2011;10:3722-30.
|
| 43. | Cilenšek I, Mankoè S, Petroviè MG, Petroviè D. GSTT1 null genotype is a risk factor for diabetic retinopathy in Caucasians with type 2 diabetes, whereas GSTM1 null genotype might confer protection against retinopathy. Dis Markers 2012;32:93-9.
|
| 44. | Thameem F, Yang X, Permana PA, Wolford JK, Bogardus C, Prochazka M. Evaluation of the microsomal glutathione S-transferase 3 (MGST3) locus on 1q23 as a Type 2 diabetes susceptibility gene in Pima Indians. Hum Genet 2003;113:353-8.
|
| 45. | Thameem F, Puppala S, Arar NH, Stern MP, Blangero J, Duggirala R, et al. Endothelial nitric oxide synthase (eNOS) gene polymorphisms and their association with type 2 diabetes-related traits in Mexican Americans. Diab Vasc Dis Res 2008;5:109-13.
|
| 46. | Casas JP, Cavalleri GL, Bautista LE, Smeeth L, Humphries SE, Hingorani AD. Endothelial nitric oxide synthase gene polymorphisms and cardiovascular disease: A HuGE review. Am J Epidemiol 2006;164:921-35.
|
| 47. | Hayden MR, Tyagi SC. Is type 2 diabetes mellitus a vascular disease (atheroscleropathy) with hyperglycemia a late manifestation? The role of NOS, NO, and redox stress. Cardiovasc Diabetol 2003;2:2.
|
| 48. | Hingorani AD. Endothelial nitric oxide synthase polymorphisms and hypertension. Curr Hypertens Rep 2003;5:19-25.
[PUBMED] |
| 49. | Hayden MR, Tyagi SC. Intimal redox stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovasc Diabetol 2002;1:3.
|
| 50. | Hayden MR, Tyagi SC. Islet redox stress: The manifold toxicities of insulin resistance, metabolic syndrome and amylin derived islet amyloid in type 2 diabetes mellitus. JOP 2002;3:86-108.
|
| 51. | Veldman BA, Spiering W, Doevendans PA, Vervoort G, Kroon AA, de Leeuw PW, et al. The Glu298Asp polymorphism of the NOS 3 gene as a determinant of the baseline production of nitric oxide. J Hypertens 2002;20:2023-7.
|
| 52. | Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 2001;104:342-5.
|
| 53. | Cromheeke KM, Kockx MM, De Meyer GR, Bosmans JM, Bult H, Beelaerts WJ, et al. Inducible nitric oxide synthase colocalizes with signs of lipid oxidation/peroxidation in human atherosclerotic plaques. Cardiovasc Res 1999;43:744-54.
|
| 54. | Makuc J, Petrovic D. No association between NOS2 and NOS3 polymorphisms and diabetic nephropathy in type 2 diabetics. Cent Eur J Biol 2012;7:404-10.
|
[Figure 1], [Figure 2]
[Table 1], [Table 2], [Table 3]
| This article has been cited by | | 1 |
The Uyghur Population and Genetic Susceptibility to Type 2 Diabetes: Potential Role for Variants inCDKAL1,JAZF1, andIGF1Genes |
|
| Manshu Song,Feifei Zhao,Longjin Ran,Mamatyusupu Dolikun,Lijuan Wu,Siqi Ge,Hao Dong,Qing Gao,Yanchun Zhai,Ling Zhang,Yuxiang Yan,Fen Liu,Xinghua Yang,Xiuhua Guo,Youxin Wang,Wei Wang | | OMICS: A Journal of Integrative Biology. 2015; 19(4): 230 | | [Pubmed] | [DOI] | | | 2 |
Association of thioredoxin reductase 2 (TXNRD2) gene polymorphisms with myocardial infarction in Slovene patients with type 2 diabetes mellitus |
|
| Stojan Kariž,Sara Mankoc,Daniel Petrovic | | Diabetes Research and Clinical Practice. 2015; | | [Pubmed] | [DOI] | | | 3 |
Oxidative stress in susceptibility to breast cancer: study in Spanish population |
|
| Patricia Rodrigues,Griselda de Marco,Jessica Furriol,Maria Mansego,Mónica Pineda-Alonso,Anna Gonzalez-Neira,Juan Martin-Escudero,Javier Benitez,Ana Lluch,Felipe J Chaves,Pilar Eroles | | BMC Cancer. 2014; 14(1): 861 | | [Pubmed] | [DOI] | |
|
|
|
|