|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2014 | Volume
: 20
| Issue : 1 | Page : 75-78 |
| |
Berardinelli-Seip syndrome type 1 in an Egyptian child
Kotb Abbass Metwalley, Hekma Saad Farghaly
Department of Pediatrics, Pediatric Endocrinology Unit, Faculty of Medicine, Assiut University, Assiut, Egypt
| Date of Web Publication | 19-May-2014 |
Correspondence Address:
Kotb Abbass Metwalley
Department of Pediatrics, Pediatric Endocrinology Unit, Faculty of Medicine, Assiut University, Assiut
Egypt
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.132762

 Abstract Abstract | | |
Berardinelli-Seip syndrome type 1 or Berardinelli-Seip congenital lipodystrophy 1 (BSCL1) is a very rare genetic disorder characterized by lipoatrophy, hypertriglyceridemia, hepatomegaly and acromegaloid features. Its prevalence in Egypt is not known. Here, we report case of a 12-year-old Egyptian boy with the clinical, metabolic and molecular genetics manifestations of BSCL1 including overt diabetes mellitus.
Keywords: Acromegaloid features, Berardinelli-Seip syndrome type 1, diabetes mellitus, hypertriglyceridemia
How to cite this article:
Metwalley KA, Farghaly HS. Berardinelli-Seip syndrome type 1 in an Egyptian child. Indian J Hum Genet 2014;20:75-8 |
| Introduction | |  |
Berardinelli-Seip syndrome or Berardinelli-Seip congenital lipodystrophy (BSCL) (MIM 269700) is a rare autosomal recessive disorder characterized by the almost total lack of subcutaneous adipose tissue, severe diabetes mellitus and no ketosis/ketonuria and insulin resistance. Other features include mental retardation; hypertrichosis; precocious puberty; nephropathy; hypertrophic cardiomyopathy. The estimated world-wide prevalence is 1 in 10 million population. [1] On the basis of mutational and haplotype analysis, BSCL families have been classified into three types BSCL1, BSCL2 and BSCLX. [1] World-wide; the prevalence of BSCL2 is somewhat more than that of BSCL1. BSCL1 is prevalent in African-American population, is the milder variety and presenting in the second or third decade of life. Garg et al., [2] first identified the gene for BSCL1 on chromosome 9q34. BSCL2 is prevalent in Portugal, Lebanon, Norway and Middle East. BSCL2 is more severe with onset in the neonatal period or early infancy. Most cases have mental retardation. The locus for BSCL2 has been identified on chromosome 11q13 by Magrι et al. [3] BSCLX families are very rare; they show evidence against co-segregation with either 9q34 or 11q13. [4] Though some cases have been reported in literatures, genetic studies of those subjects were not carried out. [5] We present the case of Berardinelli-Seip syndrome type 1in a 12 year Egyptian boy in view of its rarity.
| Case Report | | |
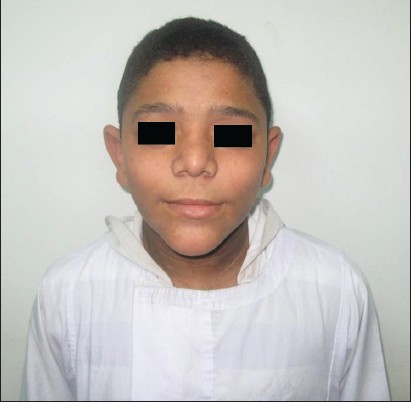
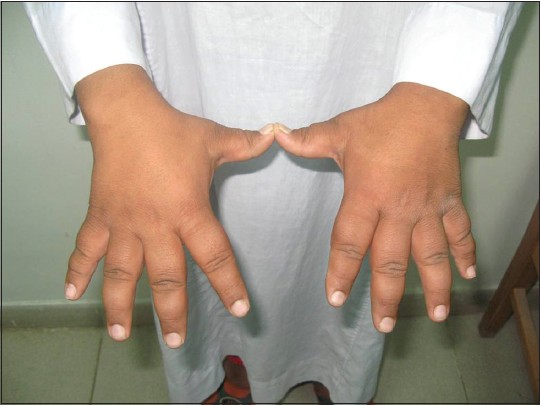
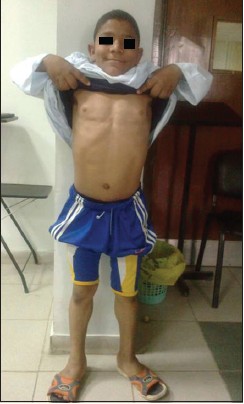
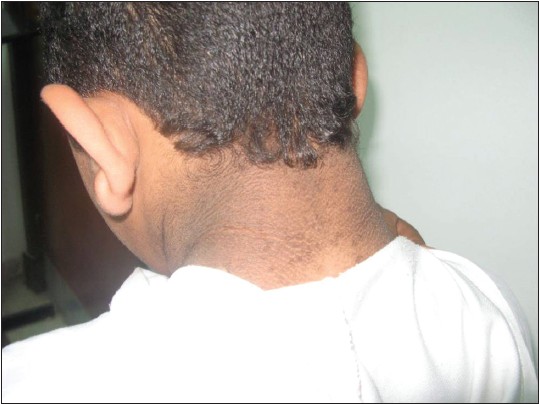
This was a case report of a 12-year-old Egyptian boy who was admitted to our institution with random blood glucose level of 450 mg/dl. He had history of polyuria and polydipsia associated with weight loss of 2 weeks duration. No other family members were affected with similar complaints. He is the younger of two siblings from a second degree consanguineous marriage. He was a normal full-term baby, with no prenatal, perinatal, or postnatal complications with up-to-date immunizations. His mental development was normal. On examination, his vital signs were normal, his weight was 47 kg (25 percentile) height: 143 cm (25 percentile) [Figure 1]. He looked older than his stated age; he had acromegaloid features (enlarged hands and feet, big ears and prominent mandible prominent orbital ridges) [Figure 2] and [Figure 3], absence of subcutaneous fat in the extremities, face and trunk; prominent pectoral and calf muscles [Figure 2] and [Figure 4]. He had acanthosis nigricans in the neck, axillae and groins [Figure 5]. Abdominal examination revealed hepatosplenomegaly. Cardiovascular evaluation was normal with no evidence of retinopathy. Investigations: Fasting blood sugar 182 mg/dl, post prandial blood sugar of 342 mg/dl, urinalysis showed +2 of glucose and absent ketones. Frequent monitoring of blood glucose revealed persistent hyperglycemia (250-350 mg/dl). Serum insulin was elevated to 68 μIU/Ml (normal up to-23 μIU/Ml) suggesting insulin resistance and simultaneous glucose was 320 mg/dl. Level of hemoglobin A1c (HbA1c) was 12.2% (4.3-5.8%). Tests for insulin antibody and anti-glutamic acid decarboxylase were negative. Leptin level was 0.25 ng/ml (normal range: (18-37.4), his lipid profile revealed cholesterol - 339 mg/dl (114-203 mg/dl), high-density lipoprotein <20 mg/dl (40- 60 mg/dl), low-density lipoprotein - 223 mg/dl (<130 mg/dl), triglycerides - 844 mg/dl (29-99 mg/dl). Complete blood picture and levels of blood urea nitrogen and creatinine were within the normal range. Liver function tests revealed bilirubin 11 umol/l (normal <20 umol/l), alanine aminotransferase 186 IU/1 (normal <50 IU/l), aspartate aminotransferase 123 IU/1 (normal <50 IU/l); alkaline phosphatase 108 IU/l, (normal <350 IU/1); albumin 38 g/l (normal range: 35-45 g/l). Viral hepatitis B and C screens were negative. Ultrasonography and magnetic resonance imaging (MRI) of the abdomen revealed an enlarged fatty liver, splenomegaly, absence of visceral fat with no evidence of portal hypertension. Cranial MRI showed normal findings. His growth hormone levels were within normal range in several measurements (0.2- 1.1 ng/mL). Serum insulin-like growth factor-1 (IGF-I) level was also normal. Other hormone levels were normal these findings ruled out the diagnosis of acromegaly. Echocardiogram was normal. In view of the typical dysmorphology, age of onset, normal mentality, hepatomegaly, diabetes mellitus, acanthosis nigricans and hypertriglyceridemia, the diagnosis of BSCL1 was kept. [6] Molecular genetic study revealed AGPAT2 mutations which are responsible for BSCL1. Our index case was started on a combined regimen of insulin, metformin and encouraged to do more physical activity. After 3 months of the above management, his blood sugar was maintained between (200-255 mg/dl), HbA1c: 8.2% and triglyceride level of 120 mg/dl.
| Discussion | | |
This disease is called. Berardinelli-Seip syndrome after Berardinelli from Brazil, who described the first patients [6] and it was confirmed by Seip from Norway in 1959. [7] The clinical and imaging features of the syndrome are mostly due to fat deficiency, diabetes, or to manifestations of secondary hyperinsulinemia, which results from the failure of the tissues to respond to insulin. The major diagnostic features of BSCL, described by Maldergem Van [8] are lipoatrophy, acromegaloid features, hepatomegaly, elevated serum triglycerides and insulin resistance (manifested as acanthosis nigricans); all of these are found in our patient. Other features include hypertrophic cardiomyopathy, mental retardation, hypertrichosis, bone cysts were not noticed in our patient.
The pathophysiology of lipodystrophies is still unknown. However, murine models of lipoatrophic diabetes revealed that primary genetic alterations in fat development resulted in diabetes and dyslipidemia. [9] Leptin deficiency, caused by the absence of adipose tissue, could be an important determinant of the metabolic abnormalities since exogenous administration or transgenic over expression of leptin has been shown to markedly improve insulin sensitivity, glycemic control, dyslipidemia and hepatic steatosis in mice. Similarly, the defect in adinopectin, another fat derived hormone, has been shown to be involved in insulin resistance. [10] Management of lipodystrophy centers on controlling the diabetes, improving the insulin resistance and reducing the triglyceride levels.
Patients with BSCL, as exemplified in our index case, are quite resistant to insulin therapy. Insulin doses as high as 1000 units daily may be required to control blood glucose levels [11] and even these doses may not suffice. Metformin has been shown to have some effect in helping patients reduce their appetite and improve and hepatic steatosis. [12] Our patient showed improvement in HbA1c on a combined regimen of insulin and metformin. Care should be taken in monitoring hepatic functions closely, as metformin is associated with hepatotoxicity. The most promising treatment for patients with congenital generalized lipodystrophy is recombinant leptin. This medication appears to improve insulin sensitivity, decrease triglyceride levels and help control energy homeostasis. This results in less food intake and lower fasting blood glucose levels as well as lower HbA1c levels. [13] Some reports show normoglycemia on leptin therapy even after other discontinuing hypoglycemic agents. [14]
Acromegaloid features which are characteristic and consist of prognathism, enlarged hands/feet and big ears are all thought to be a result of insulin cross-reacting with IGF-1 receptors. In addition, our index case had hepatic steatosis which result in a mild transaminitis as triglycerides deposit into the liver secondary to a paucity of generalized fat tissue. [15] As a consequence of near-total loss of body fat, serum levels of adipocytokines such as leptin and adiponectin are low. [16] In cotrast, the morphologic and functional study of the muscles in patients BSCL suggests that the increase in muscle mass results from hyperplasia and not from hypertrophy. [17] Patients with BSCL must have a multidisciplinary follow-up. They should consume a low-fat diet, with reduction of saturated fats and cholesterol intake. It is also important to practice daily physical activity. [3]
BSCL type 1 seems to be less severe; with some cases of type 2 resulting in premature death, which can occur as early as the 1 st year of life. Approximately 80% of individuals with mutations in BSCL2 have mild-to-moderate intellectual impairment, whereas only 10% of individuals with mutations in AGPAT2 have intellectual impairment. The mutations in AGPAT2 may cause congenital lipodystrophy by inhibiting/reducing triacylglycerol synthesis and storage in adipocytes. It is also likely that reduced AGPAT2 activity could increase tissue levels of lysophosphatidic acid, which may negatively affect adipocyte functions. [18] It has been shown that lipodystrophy is induced by a compound similar to cachectin [19] (tumor necrosis factor). This has powerful inhibitory effects on lipoprotein lipase and causes fat depletion and hyperlipidemia when injected in animals.
Genetic study is helpful in determination of the prognosis, since BSCL1 is a milder disease with lower occurrences of mental retardation and premature death than BSCL2. Recurrence risk is 25%, so chorionic villus sampling at 9-12 weeks is recommended for prenatal diagnosis in at risk families. [8]
| Conclusions | | |
Berardinelli-Seip syndrome type 1 is a rare disease which causes important metabolic abnormalities, which can complicate and have a fatal out come if optimal therapeutic, preventive measures and a multidisciplinary follow-up are not adopted. Pediatricians should keep this possibility in mind while evaluating patients with similar clinical features.
| References | | |
| 1. | Babu P, Sharma R, Jayaseelan E, Appachu D. Berardinelli-Seip syndrome in a 6-year-old boy. Indian J Dermatol Venereol Leprol 2008;74:644-6. 
[PUBMED]  |
| 2. | Garg A, Wilson R, Barnes R, Arioglu E, Zaidi Z, Gurakan F, et al. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab 1999;84:3390-4.
|
| 3. | Magré J, Delépine M, Khallouf E, Gedde-Dahl T Jr, Van Maldergem L, Sobel E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet 2001;28:365-70.
|
| 4. | Mandal K, Aneja S, Seth A, Khan A. Berardinelli-Seip congenital lipodystrophy. Indian Pediatr 2006;43:440-5.
|
| 5. | Garg A. Lipodystrophies. Am J Med 2000;108:143-52.
|
| 6. | Berardinelli W. An undiagnosed endocrinometabolic syndrome: Report of 2 cases. J Clin Endocrinol Metab 1954;14:193-204.
|
| 7. | Seip M. Lipodystrophy and gigantism with associated endocrine manifestations. A new diencephalic syndrome? Acta Paediatr 1959;48:555-74.
|
| 8. | Maldergem Van L. Berardinelli-Seip congenital lipodystrophy. Orphanet encyclopedia. Available from: http://www.orpha.net/data/patho/GB/ukberard.pdf. [Last cited on 2012 Jun 2012].
|
| 9. | Van Maldergem L, Magré J, Khallouf TE, Gedde-Dahl T Jr, Delépine M, Trygstad O, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet 2002;39:722-33.
|
| 10. | Bhayana S, Hegele RA. The molecular basis of genetic lipodystrophies. Clin Biochem 2002;35:171-7.
|
| 11. | Meyer L, Hadjadj S, Guerci B, Delbachian I, Ziegler O, Drouin P. Lipoatrophic diabetes mellitus treated by continuous subcutaneous insulin infusion. Diabetes Metab 1998;24:544-6.
|
| 12. | Lillioja S, Bogardus C. Obesity and insulin resistance: Lessons learned from the Pima Indians. Diabetes Metab Rev 1988;4:517-40.
|
| 13. | Pardini V, Victoria I, Rocha S. Metformin improves metabolic control in subjects with Congenital Generalized Lipoatrophic Diabetes. In: 57th Annual Scientific Sessions, Boston − Massachusetts. Diabetes. 1997;46:160.
|
| 14. | Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med 2002;346:570-8.
|
| 15. | Gomes KB, Pardini VC, Fernandes AP. Clinical and molecular aspects of Berardinelli-Seip Congenital Lipodystrophy (BSCL). Clin Chim Acta 2009;402:1-6.
|
| 16. | Jaquet D, Khallouf E, Lévy-Marchal C, Czernichow P. Extremely low values of serum leptin in children with congenital generalized lipoatrophy. Eur J Endocrinol 1999;140:107-9.
|
| 17. | Figueiredo-Filho PP, Val AC, Diamante R, Cunha CF, Norton RC, Lamounier JA, et al. Congenital generalized lipodystrophy. J Pediatr (Rio J) 2004;80:333-6.
|
| 18. | Agarwal AK, Arioglu E, De Almeida S, Akkoc N, Taylor SI, Bowcock AM, et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet 2002;31:21-3.
|
| 19. | Kher AS, Lahiri KR, Jain MK, Shah MD. Congenital lipodystrophy with defective leucocyte function (a case report). J Postgrad Med 1990;36:48-50.
[PUBMED] |
[Figure 1], [Figure 2], [Figure 3], [Figure 4], [Figure 5]
| This article has been cited by | | 1 |
Berardinelli–Seip syndrome type 2 – An Egyptian child |
|
| Rabah M. Shawky,Radwa Gamal,Neveen S Seifeldin | | Egyptian Journal of Medical Human Genetics. 2014; | | [Pubmed] | [DOI] | |
|
|
|
|