|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2014 | Volume
: 20
| Issue : 2 | Page : 199-202 |
| |
First report of c. 1499G>C mutation in a 6-month-child with cystic fibrosis
Abbas Sahami1, Nourkhoda Sadeghifard2, Alireza Monsef3, Hadi Peyman4
1 Department of Medical Genetic and Embryology, Faculty of Medicine, Ilam University of Medical Sciences, Ilam, Iran
2 Department of Microbiology, Faculty of Medicine; Clinical Microbiology Research Center, Ilam University of Medical Sciences, Ilam, Iran
3 Department of Pathology, Hamadan University of Medical Sciences, Hamadan, Iran
4 Research Center for Prevention of Psychosocial Injuries, Ilam University of Medical Sciences, Ilam, Iran
| Date of Web Publication | 14-Oct-2014 |
Correspondence Address:
Nourkhoda Sadeghifard
Department of Microbiology, Faculty of Medicine, Ilam University of Medical Sciences, Pazhuhesh BLVD, Ilam
Iran
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.142911

 Abstract Abstract | | |
So far, more than 1800 mutations identified in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. In this case report, we presented first report of c. 1499G>C mutation in a 6-month-old girl with cystic fibrosis (CF) diagnosis. A 6-month-old girl with weakness and meconium Ileus referred to the pediatric clinic in Ilam, in the west of Iran. Patient's skin was dark and suffered from bronchiectasis. The sweat test was performed, and the concentration of chloride and sodium in patient's sweat was 130-135 mmol/L and 125-128 mmol/L, respectively. The exon 10 mutation analysis of a CF patient was performed. CFTR mutation analysis revealed the identification of 2 mutations in patient, the mutations were p.F508del (ΔF508) and c. 1499G>C (cd500), respectively. The mutation c. 1499G>C (cd500) were found for the first time in the world. Assessing this mutation in future study and genetic investigation is recommended.
Keywords: c. 1499G>C, cystic fibrosis, cystic fibrosis transmembrane conductance regulator, direct sequencing, Iran, mutations
How to cite this article:
Sahami A, Sadeghifard N, Monsef A, Peyman H. First report of c. 1499G>C mutation in a 6-month-child with cystic fibrosis. Indian J Hum Genet 2014;20:199-202 |
How to cite this URL:
Sahami A, Sadeghifard N, Monsef A, Peyman H. First report of c. 1499G>C mutation in a 6-month-child with cystic fibrosis. Indian J Hum Genet [serial online] 2014 [cited 2016 Aug 23];20:199-202. Available from: http://www.ijhg.com/text.asp?2014/20/2/199/142911 |
| Introduction | |  |
The cystic fibrosis transmembrane conductance regulator (CFTR) is a phosphorylation and nucleotide regulated chloride (Cl) channel. The incidence of cystic fibrosis (CF) (MIM# 219700) is very varies in different areas for example in Australia is 1:248 000 live births. [1] However, the incidence of CF in many studies was reported 1:2500 live births and 0.4 in carriers. [2] In US, screening for CF made that more infants are being diagnosed in every state. [3]
Cystic fibrosis is the inheritance an autosomal recessive. [4] CF disease caused by mutations in CFTR gene that localized on the long arm of chromosome number 7 (7q31, 2). This gene has 27 exon and expressed protein by this gene had a 1480 amino acid, [5],[6] which encodes a cyclic adenosine monophosphate-dependent Cl channel that is found at the apical membrane of epithelial cells, including respiratory epithelia and submucosal glands, exocrine pancreas, liver, sweat ducts, and the reproductive tract. CFTR is a member of the adenosine triphosphate-binding cassette membrane transporter superfamily that includes proteins such as the multiple drug resistance protein and bacterial periplasmic permeases. [7],[8]
| Case Report | | |
A 6-month-old girl with weakness and meconium Ileus referred to the pediatric clinic in Ilam, in the west of Iran [Figure 1]. Weight and height of the patient was 6 kg and 55 cm, respectively. Patient's skin was dark, and their parents stated that the child had a weight loss. The child was also suffered from bronchiectasis. There are no evidence for edema, hypoproteinemia, liver and gallbladder problems, rectal prolapsed or Finger Clubbing [Table 2].
The pediatric specialist requested sweet test. By using Macroduct method (Wescor, Logan, UT), the sweat test was performed twice. The concentration of (Cl) and sodium in patient's sweat was 130-135 mmol/L and 125-128 mmol/L, respectively. The consent was performed from parents to getting sweat test and blood sample. A sample of 4 ml blood was taken for desoxyribonucleic acid (DNA) extraction.
Desoxyribonucleic acid was extracted by using salting out method. All samples were sequenced in both the forward and reverse direction using the same primers used in the polymerase chain reaction (PCR) reactions. The conditions used for amplification were as follows: 5 min at 94°C; 30 cycles of 30 s at 94°C followed by 30 s at annealing temperature, and 30 s at 72°C; and a final extension step for 5 min at 72°C. The primers used in this study were synthesized by Operon (Metabion, Germany). The sequences of primers, listed in 5'-3'-direction, are shown in [Table 1].
The sequencing reactions were vacuums purified with the QIAquick PCR purification kit and analyzed on an ABI 3130 Gene Analyzer (Applied Biosystems, USA). The sequences were compared with the wild-type CFTR nucleotide sequence using DNA sequencing analysis v5.2 software (Applied Biosystems, USA). Identified sequence changes were confirmed by direct DNA sequencing in the reverse direction.
After comprehensive genetic analysis of exon 10, there was a mutation at the position c. 1499G>C to be heterozygous (GLY >ALA) [Figure 2] and [Figure 3], [Table 3]. The child has also a ΔF508 mutation as a heterozygous [Figure 4] and [Table 3].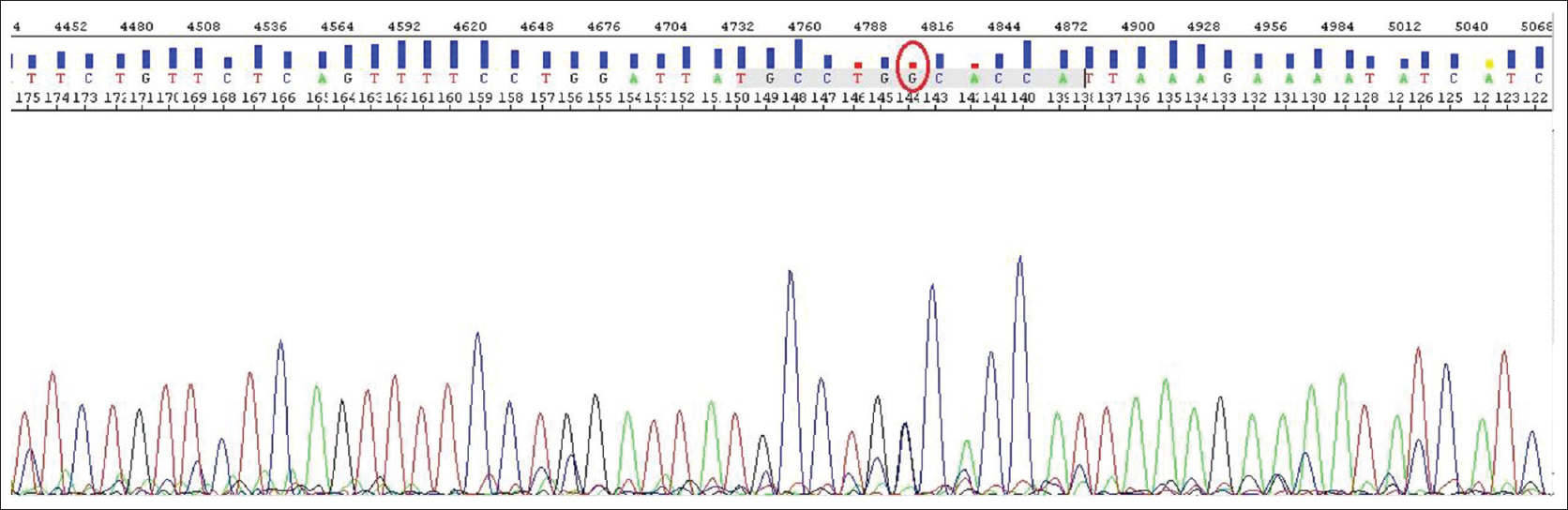 | Figure 2: The heterozygote c.1499 mutation (G>C). The sequence resulted from exon 10 sequencing which heterozygote CD500 mutation has created in 1499 place
Click here to view |
 | Figure 3: Identification of mutation in Exon 10 of cystic fibrosis transmembrane conductance regulator gene
Click here to view |
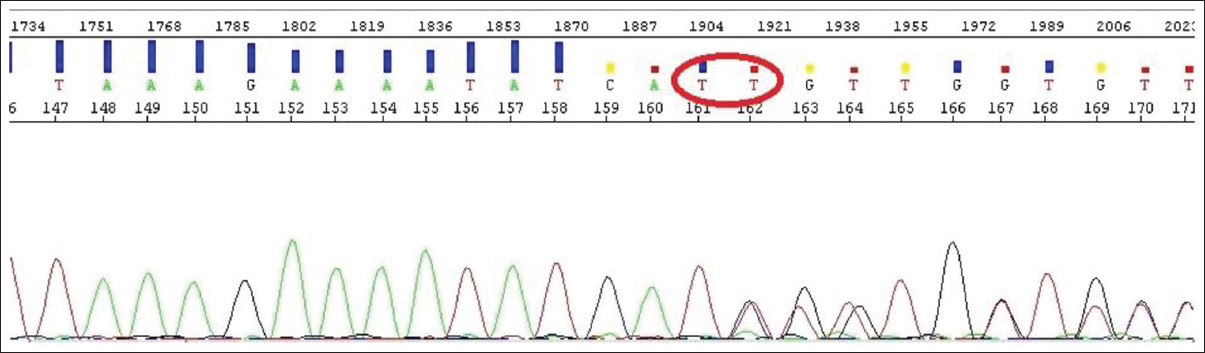 | Figure 4: The sequence resulted from sequencing process which ΔF508 mutation is seen as a heterozygote type. In this sequence, disruption in arrangement of the peaks demonstrates that one of the transmitted hereditary genes from parents to the child had been deficit
Click here to view |
| Conclusion | | |
The investigation to better recognition of CFTR mutations not only is useful in clinical recognition of CF, but it further strengthens the available genetical knowledge. These findings are also beneficent in pre-marriage consultant protocols, and they lead to easier carrier identification. The results in this report and other related papers indicate a high rate of heterogeneity in CFTR mutations.
Up to 2012, more than 1800 (Cystic Fibrosis Mutation Database, http://www.genet.sickkids.on.ca/cftr/.) mutations in the CFTR gene were detected and reported in Caucasian or- non-Caucasian. The type and distribution of mutations varies widely between different countries and/or ethnic groups.
According to the geographical and ethnic origin of patients, there are many variations in frequency and prevalence of these mutations. Many of these mutations are rare, and some of them have been reported in only one family. [5]
Clinical profile and laboratory findings and genotype analysis
A patient with a sign of CF with consanguine parents (Degree of 3) in Ilam province (west of Iran) [Figure 1] has been studied. The patient was analyzed by the help of sequencing. She was a 6 month baby and found with acute respiratory disorders symptoms, growth retardation and abnormal stool. The analysis shows the c. 1499G>C and ΔF508 mutation on exon 10. The mutation of c. 1499G>C is being reported for the first time in this case report. The patient had symptoms of meconium ileus and gastrointestinal obstruction and was found to be conveying heterozygote ΔF508 mutation. The sign of liver disorders, hypoproteinemia, rectal prolapse and mellitus diabetes were not found in the patient.
In Middle East populations, there is no complete information about the CF disease and limited studies have been conducted. Hence, it seems that ethnic and tribal relationships in these areas have led to frequent native mutations. For prevention of increase CF cases, many of preventive programs like Establishment of screening tests for diagnostic of carrier statues must be done special in developing countries like Iran. Further studies to identify frequent and native mutations in these countries are recommended.
| Acknowledgment | | |
The author would like to appreciate the family of patient whom this study would not be accomplished without their sincere support.
| References | | |
| 1. | Brooks DA, Gibson GJ, Karageorgos L, Hein LK, Robertson EF, Hopwood JJ. An index case for the attenuated end of the mucopolysaccharidosis type VI clinical spectrum. Mol Genet Metab 2005;85:236-8.  |
| 2. | Kanavakis E, Efthymiadou A, Strofalis S, Doudounakis S, Traeger-Synodinos J, Tzetis M. Cystic fibrosis in Greece: Molecular diagnosis, haplotypes, prenatal diagnosis and carrier identification amongst high-risk individuals. Clin Genet 2003;63:400-9. |
| 3. | Padman R, Flathers K, Passi V. Cystic fibrosis infant care challenges in diagnosis and management in the era of newborn screening. Del Med J 2012;84:149-55. |
| 4. | Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998;95:1005-15. |
| 5. | Sahami A, Alibakhshi R, Ghadiri K, Sadeghi H. Mutation Analysis of Exons 10 and 17a of CFTR Gene in Patients with Cystic Fibrosis in Kermanshah Province, Western Iran. J Reprod Infertil 2014;15:49-56. |
| 6. | Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989;245:1059-65. |
| 7. | Alibakhshi R, Kianishirazi R, Cassiman JJ, Zamani M, Cuppens H. Analysis of the CFTR gene in Iranian cystic fibrosis patients: Identification of eight novel mutations. J Cyst Fibros 2008;7:102-9. |
| 8. | Dorwart M, Thibodeau P, Thomas P. Cystic fibrosis: Recent structural insights. J Cyst Fibros 2004;3 Suppl 2:91-4. |
[Figure 1], [Figure 2], [Figure 3], [Figure 4]
[Table 1], [Table 2], [Table 3]
|