|
 
 |
| CASE REPORT |
|
| Year : 2016 | Volume
: 17
| Issue : 2 | Page : 64-67 |
|
Caudal regression syndrome with a solitary kidney: A case report and review of the literature
Abdulkadir Abubakar1, Babagana Mustapha Abubakar2
1 Department of Surgery, Bayero University/Aminu Kano Teaching Hospital, Kano, Nigeria
2 Department of Surgery, Federal Medical Centre, Nguru, Yobe, Nigeria
| Date of Web Publication | 16-Nov-2016 |
Correspondence Address:
Abdulkadir Abubakar
Department of Surgery, Bayero University/Aminu Kano Teaching Hospital, Kano, PMB 3452, Kano State
Nigeria
 Source of Support: None, Conflict of Interest: None  | Check |
DOI: 10.4103/1595-1103.194213

Caudal regression syndrome (CRS) is a rare, congenital anomaly that is often complicated by a neurogenic bladder. Congenital anomalies of the kidney and urinary tract in aforesaid patient present an added challenge in the management. The aim and objective of this study is to report a rare case of CRS with neurogenic bladder and right solitary hydronephrotic kidney. The patient was a 3-year-old female infant with burdensome lower urinary tract symptoms and congenital back and lower limbs deformities. Her examination unveiled vertebra and lower limbs anomalies. The computed tomography scan showed lumbosacrococcygeal agenesis with a right solitary hydronephrotic kidney. She was treated for urinary tract infection at presentation, after which cystometry confirmed high-pressure neurogenic bladder. She did well to the initial continuous bladder drainage and the ensuing oxybutynin with clean intermittent catheterization. Instantaneous management of the bladder dysfunction prevents potentially permanent solitary renal dysfunction and the secondary bladder malfunction in a patient with CRS and neurogenic bladder. Keywords: Caudal dysplasia, caudal dysplasia sequence, congenital sacral agenesis, lumbosacral agenesis, sacral regression, solitary kidney
How to cite this article:
Abubakar A, Abubakar BM. Caudal regression syndrome with a solitary kidney: A case report and review of the literature. Niger J Surg Res 2016;17:64-7 |
How to cite this URL:
Abubakar A, Abubakar BM. Caudal regression syndrome with a solitary kidney: A case report and review of the literature. Niger J Surg Res [serial online] 2016 [cited 2018 Jul 21];17:64-7. Available from: http://www.njsrjournal.org/text.asp?2016/17/2/64/194213 |
| Introduction | |  |
Congenital anomaly of the kidney and urinary tract (CAKUT) is a momentous source of chronic kidney disease and end-stage renal diseases that could result in mortality.[1] In a child with caudal regression syndrome (CRS), the musculoskeletal anomalies have a serious impact on the body image, and in the patient's care, it may overpass the more life-threatening CAKUT. The index patient presented with these musculoskeletal anomalies, neurogenic bladder, and right solitary hydronephrotic kidney. A written signed consent from the patient's parent and clearance from the Hospital Ethics Committee were obtained to report this case with its distinctive management challenges.
| Case Report | | |
The patient was 3-year-old who first presented when she was 2 weeks with difficulty in passing urine that was characterized by frequent scanty voiding with excessive crying. She had episodes of suprapubic swelling that receded with nasogastric tube drainage of urine from the bladder by pediatricians before her presentation. There was no history indicative of upper tract symptoms and no fever. The mother observed swelling at the back of the patient with lower limbs' deformities since birth. There was no history indicative of other congenital anomalies.
The pregnancy was booked with regular antenatal follow-up. The fetal ultrasound, however, revealed no anomaly. Mother had no history of exposure to teratogens although she was on treatment for pregnancy-induced hypertension; her blood pressure was controlled with Aldomet. The labor was at term and the delivery via spontaneous vaginal delivery; all were supervised and uneventful. The patient cried immediately after birth, weighed 2.8 kg, and passed meconium within 6 hours after delivery.
Mother is a 40-year-old para (8 + 1), not a known diabetic, and had no other significant medical condition. Mother did neither smoke cigarette nor drink alcohol and no history of congenital anomalies in the family.
On examination, the patient was not pale and afebrile to touch. She was in Buddha posture and had short webbed neck, short trunk with kyphosis along the lumbar vertebral spine. She had an underdeveloped pelvis, dysplastic hip joint, and hypoplastic gluteal region. Her lower limbs were in flexion with webbing at the knee joints and varus deformity bilaterally. The umbilical stump was low set [Figure 1] and [Figure 2], no palpable organomegaly. Her vulva had normal urethral and vaginal openings. There was a considerable skin excoriation at the perianal region. The anal opening was sited anteriorly to the typical position and the sphincteric tone was hypotonic.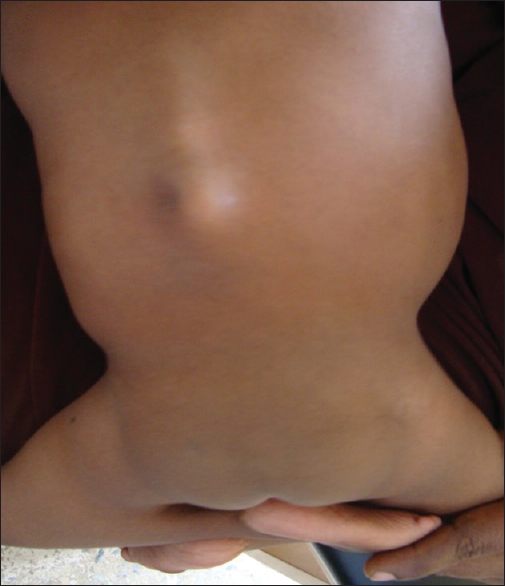 | Figure 1: The patient's back showing kyphosis along the lumbar vertebral spine, underdeveloped pelvis, dysplastic hip joint, and hypoplastic gluteal region
Click here to view |
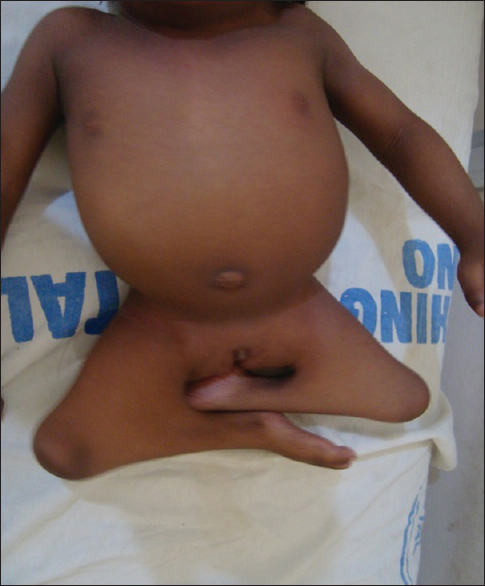 | Figure 2: Patients Budha posture, short trunk, flexes deformity with webbing at the knee joints and low set umbilicus
Click here to view |
At presentation, she was managed as a medical and social emergency. The urine retention was promptly relieved and indwelling 6Fr catheter left in situ. The initial urgent urea, electrolytes, and creatinine affirmed normal electrolytes but elevated urea of 8.1 mmol that normalized on the 3rd day. Her urine microscopy, culture, and sensitivity yielded Klebsiella spp. sensitive to augmentin and she was duly treated. The full blood count and differentials revealed leukocytosis and her genotype was AA. The computed tomography (CT) revealed lumbosacrococcygeal agenesis with a right hydronephrotic solitary kidney [Figure 3] and [Figure 4]. Her cystometry confirmed high-pressure neurogenic bladder [Figure 5]. She did well to the initial continuous bladder drainage while the mother was trained on clean intermittent catheterization (CIC). The abdominal ultrasound afterwards revealed a reduction in the severity of hydronephrosis with kidney size of 7.8 cm × 3.8 cm. She was then maintained on oxybutynin and 3 hourly CIC. The social emergency was managed through the parent counseling. The patient did well clinically and the repeated urine microscopy, culture, and sensitivity plus serum urea, electrolytes and creatinine were normal. Serial abdominopelvic ultrasound revealed only moderate residual hydronephrosis with good corticomedullary differentiation, kidney size of 7.8 cm × 3.8 cm and she has been on follow-up.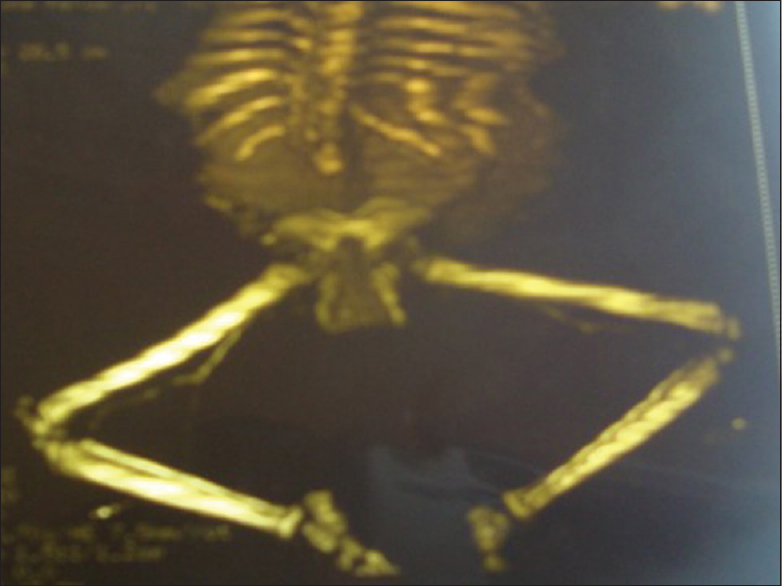 | Figure 3: Computed tomography sonogram, showing the lumbosacral agenesis (total sacrococcygeal and lower three lumbar agenesis with the last formed vertebral segment resting on the fused ilia) with pubic diastasis
Click here to view |
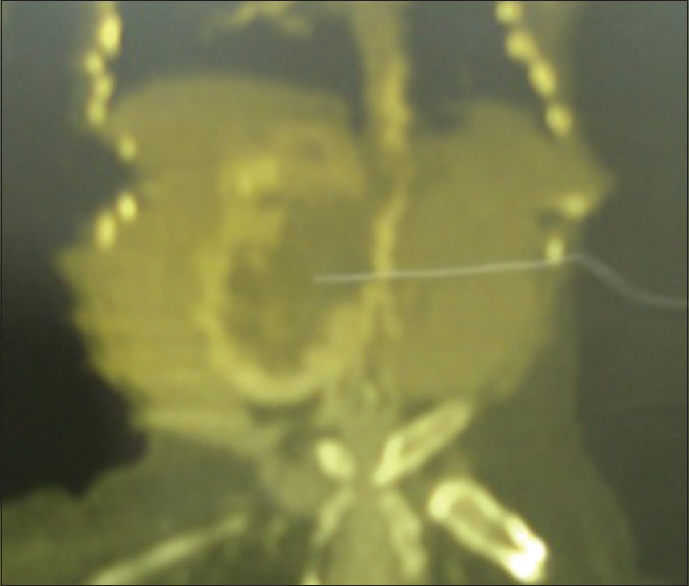 | Figure 4: Computed tomography sonogram, showing a solitary right hydronephrotic kidney
Click here to view |
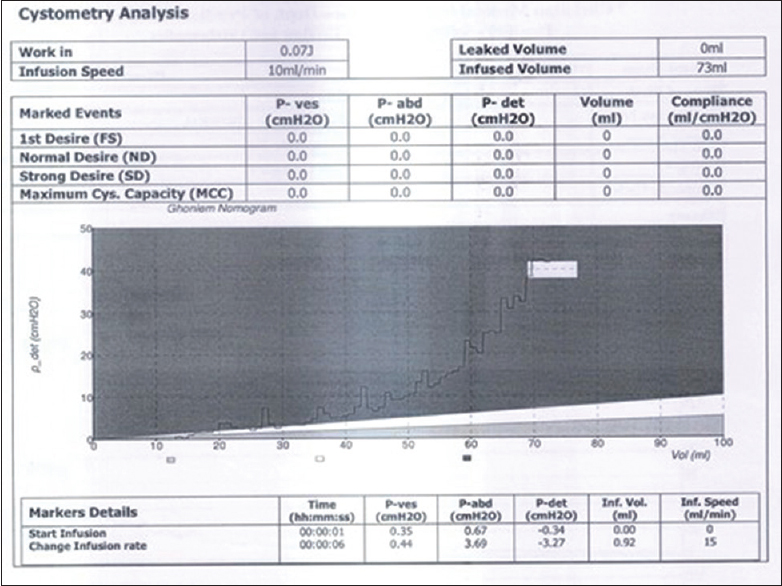 | Figure 5: Cystometry at infusion rate of 10 ml per min shows detrusor pressure rising above 20 cm H2O at 58 ml infusion and above 40 cm H20 at 70 ml infusion at which she leaked against her expected capacity of 120 ml
Click here to view |
| Discussion | | |
The kidney development is initiated at around the 5th week of intrauterine life and results from a complex interplay between the ureteric bud (UB) and the metanephric mesenchyme (MM). The misstep of the complementary induction between MM and UB upsetting normal renal development manifests in different forms of CAKUT.[2] These anomalies occur approximately in 1 of 600 births [3] and are customarily aftermath of genetic and environmental insults. The environmental insults include drugs administered during pregnancy;[4] however, our patient's mother took none of the cited drugs; she only had Aldomet for pregnancy-induced hypertension with the routine antenatal hematinic. Other risk factors for CAKUT include small-for-gestational age and maternal diabetic mellitus.[5] In the index case, however, the maternal blood glucose level and the patient's birth weight were normal.
The genes under suspicion in nonsyndromic CAKUT anomalies include PAX2, HNF1B, and DSTYK;[6] heterozygous mutation in these genes is culpable in 10%–20% cases.[7],[8] The most critical in the spectrum of these anomalies is renal agenesis (RA). One-tenth of RAs are familial,[9] inherited largely via autosomal dominant with incomplete penetrance, and a few have autosomal recessive and X-linked hereditary.[9] It was reported that over one-fifth of newborn with chromosomal abnormalities have RA;[10] this justified the holistic approach to the evaluation of this patients at birth. The reported incidence indicated the male preponderance of RA, as well as left-sided predominance;[11] index patient is female with left-sides agenesis. Unilateral RA (URA) has an estimated worldwide incidence of 1 in approximately 2000 births and could develop from (prenatal) involution of a multicystic dysplastic kidney (5% cases)[12] or renal hypodysplasia.[13] Approximately 33% of these patients have ipsilateral CAKUT, including vesicoureteral reflux and pelviureteric junction obstruction, megaureter, or a duplex kidney;[13] the index patient had hydroureteronephrosis with neurogenic bladder dysfunction
CRS has an incidence of 1 to 0.2.5 per 100,000 births [14] and hence less common than RA. Although its precise etiology is unknown, studies suggested abnormal gastrulation leading to a disturbed mesodermal migration. This occurs around the 3rd to 7th week of intrauterine life, approximately same timing with RA. The timing of the exposed insults with the aberration of gastrulation plus the common environmental and genetic risk factors with RA explained possible common etiological linkage of the VACTERL associations. The severity of such insults may or may not correlate with the degree of the anomaly. The identified genetic risks for CRS are mutations involving HOXD13, CYP26A1, 18p11.2, and VANGL1 genes (also identified in CAKUT). Environmental factors are believed to play a role as evidence by 250-time incidence seen with overt Type 1 diabetes around conception [15],[16] (a known risk for the CAKUT). Prenatal diagnosis is possible in both CAKUT and URA, with antenatal ultrasound which will reveal short crown-rump length in the first trimester, and vertebra with spinal anomalies in the subsequent trimesters in CRS.[17] The solitary kidney may be picked by fetal ultrasound. These were missed in the index patient, and therefore, the opportunity to commence apt prenatal and immediate neonatal care was missed. The patient's cystometry confirmed high-pressure neurogenic bladder seen in up to 61% cases of CRS.[18]
The CT scan showed lumbosacrococcygeal agenesis (Renshaw Type IV);[19] the worst in this classification. The treatment for the bladder dysfunction and the UTI in the patient improved the renal function as in most cases of CRS with neurogenic bladder.[20] She was maintained on oxybutynin [20],[21],[22] and 3 hourly CIC by the mother,[23] which are the gold standard treatments in most centers.[24],[25]
The social challenge was managed through stepwise parental counseling commensurate with their level of understanding. This provided the parents with the required pandect overview of the patient's anomalies, diagnosis, and treatments alongside the restraints in the management.[26]
| Conclusion | | |
Early management of the bladder dysfunction prevents potentially irreparable solitary renal dysfunction and the secondary bladder damage in a patient with CRS. The management should preferably commence prenatally following fetal ultrasound diagnosis. An interdisciplinary approach in the management and subsequent follow-up is fundamental.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| References | | |
| 1. | Sanna-Cherchi S, Ravani P, Corbani V, Parodi S, Haupt R, Piaggio G, et al. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int 2009;76:528-33.  |
| 2. | Schedl A. Renal abnormalities and their developmental origin. Nat Rev Genet 2007;8:791-802. |
| 3. | Wiesel A, Queisser-Luft A, Clementi M, Bianca S, Stoll C; EUROSCAN Study Group. Prenatal detection of congenital renal malformations by fetal ultrasonographic examination: An analysis of 709,030 births in 12 European countries. Eur J Med Genet 2005;48:131-44. |
| 4. | Schreuder MF, Bueters RR, Huigen MC, Russel FG, Masereeuw R, van den Heuvel LP. Effect of drugs on renal development. Clin J Am Soc Nephrol 2011;6:212-7. |
| 5. | Tran S, Chen YW, Chenier I, Chan JS, Quaggin S, Hébert MJ, et al. Maternal diabetes modulates renal morphogenesis in offspring. J Am Soc Nephrol 2008;19:943-52. |
| 6. | Sanna-Cherchi S, Sampogna RV, Papeta N, Burgess KE, Nees SN, Perry BJ, et al. Mutations in DSTYK and dominant urinary tract malformations. N Engl J Med 2013;369:621-9. |
| 7. | Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: Results of the ESCAPE study. J Am Soc Nephrol 2006;17:2864-70. |
| 8. | Thomas R, Sanna-Cherchi S, Warady BA, Furth SL, Kaskel FJ, Gharavi AG. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol 2011;26:897-903. |
| 9. | Sanna-Cherchi S, Caridi G, Weng PL, Scolari F, Perfumo F, Gharavi AG, et al. Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr Nephrol 2007;22:1675-84. |
| 10. | Garne E, Dolk H, Loane M, Boyd PA; EUROCAT. EUROCAT website data on prenatal detection rates of congenital anomalies. J Med Screen 2010;17:97-8. |
| 11. | Schreuder MF. Unilateral anomalies of kidney development: Why is left not right? Kidney Int 2011;80:740-5. |
| 12. | Schreuder MF, Westland R, van Wijk JA. Unilateral multicystic dysplastic kidney: A meta-analysis of observational studies on the incidence, associated urinary tract malformations and the contralateral kidney. Nephrol Dial Transplant 2009;24:1810-8. |
| 13. | Westland R, Schreuder MF, Ket JC, van Wijk JA. Unilateral renal agenesis: A systematic review on associated anomalies and renal injury. Nephrol Dial Transplant 2013;28:1844-55. |
| 14. | Sen KK, Patel M. Caudal regression syndrome. Med J Armed Forces India 2007;63:178-9. |
| 15. | Chan BW, Chan KS, Koide T, Yeung SM, Leung MB, Copp AJ, et al. Maternal diabetes increases the risk of caudal regression caused by retinoic acid. Diabetes 2002;51:2811-6. |
| 16. | Hardardottir H, Gabbe SG, Niebyl JR, Simpson JL, Landon MB, Galan HL, et al., editors. Obstetrics Normal and Problem Pregnancies. 6 th ed. Philadelphia: Elsevier Saunders; 2012. p. 1312. |
| 17. | Negrete LM, Chung M, Carr SR, Tung GA. In utero diagnosis of caudal regression syndrome. Radiol Case Rep 2015;10:1049. |
| 18. | Torre M, Buffa P, Jasonni V, Cama A. Long-term urologic outcome in patients with caudal regression syndrome, compared with meningomyelocele and spinal cord lipoma. J Pediatr Surg 2008;43:530-3. |
| 19. | Rensahw TS. Sacral agenesis: A classification and review of 23 cases. J Bone Joint Surg 1978;60:373-83. |
| 20. | Bauer SB, Joseph DB. Management of the obstructed urinary tract associated with neurogenic bladder dysfunction. Urol Clin North Am 1990;17:395-406. |
| 21. | Baskin LS, Kogan BA, Benard F. Treatment of infants with neurogenic bladder dysfunction using anticholinergic drugs and intermittent catheterisation. Br J Urol 1990;66:532-4. |
| 22. | Fernandes ET, Reinberg Y, Vernier R, Gonzalez R. Neurogenic bladder dysfunction in children: Review of pathophysiology and current management. J Pediatr 1994;124:1-7. |
| 23. | Joseph DB, Bauer SB, Colodny AH, Mandell J, Retik AB. Clean, intermittent catheterization of infants with neurogenic bladder. Pediatrics 1989;84:78-82. |
| 24. | Kasabian NG, Bauer SB, Dyro FM, Colodny AH, Mandell J, Retik AB. The prophylactic value of clean intermittent catheterization and anticholinergic medication in newborns and infants with myelodysplasia at risk of developing urinary tract deterioration. Am J Dis Child 1992;146:840-3. |
| 25. | Tekgül S, Riedmiller H, Gerharz E, Hoebeke P, Kocvara R, Nijman R, et al. Guidelines on. Update; March, 2008. |
| 26. | Gilmore JV. Parent counseling: Theory and application. J Educ 1971;154:40-9. |
[Figure 1], [Figure 2], [Figure 3], [Figure 4], [Figure 5]
|








 Search Pubmed for
Search Pubmed for