|
 
 |
| REVIEW ARTICLE |
|
| Year : 2017 | Volume
: 12
| Issue : 2 | Page : 66-73 |
|
Comorbid depression in sickle cell disease: An overview of determinants and need for early detection
John C Aneke, Chide E Okocha
Department of Haematology and Blood Transfusion, Nnamdi Azikiwe University, Nnewi Campus, Anambra State, Nigeria
| Date of Web Publication | 24-May-2018 |
Correspondence Address:
John C Aneke
Transfusion, Nnamdi Azikiwe University, PMB 5001, Nnewi Campus, Anambra State
Nigeria
  | Check |
DOI: 10.4103/summ.summ_11_17 
Sickle cell disease (SCD) is a chronic illness, characterized by periods of painful crises, frequent hospitalizations, and multiorgan dysfunction; patients are frequently exposed to diverse psychological stressors and insults which adversely impact on overall quality of life (QOL) and survival. The following key words: “sickle cell disease,” “psychological dysfunction,”, “psychopathology,” “co-morbid depression,” “quality of life,” “disease severity,” “treatment,” and “clinical outcome” were used for literature search on PubMed, PubMed Central, Google Scholar, African Index Medicus, and Scopus database sources. No limitation as to the year of publication was applied and the oldest paper retrieved was published in 1989. The search was restricted to depression occurring in the background of SCD and publications in English language. The studies retrieved dealt mainly on the epidemiology, etiopathogenesis, and treatment of co-morbid depression in SCD, whereas papers dealing primarily with depression not related to SCD were rejected. All papers identified were assessed by the authors with a view to highlighting the prevalence and effect of depression on the clinical course of SCD. Comorbid depression was shown to constitute a significant burden in subjects living with SCD; the prevalence of which increases with increasing disease severity. In affected SCD patients, it has adverse effects on QOL and disease course. The need for early detection of comorbid depression in patients with SCD is hereby emphasized, with a view to instituting appropriate treatment geared toward ameliorating its adverse effect on disease morbidity.
Keywords: Psychopathology, quality of life, sickle bone pain crises, sickle cell disease
How to cite this article:
Aneke JC, Okocha CE. Comorbid depression in sickle cell disease: An overview of determinants and need for early detection. Sudan Med Monit 2017;12:66-73 |
| Introduction | |  |
Sickle cell disease (SCD) is a chronic multisystem disorder characterized by a single point substitution in the beta hemoglobin gene. It is one of the most common genetic disorders globally, having high prevalence rates in Africa, Middle East, Asia, and parts of America, (particularly among the African-American subpopulation).[1],[2],[3] The hallmark of the disease includes red cell sickling under conditions of reduced oxygen tension, with resultant ischemia, vascular dysfunction, and end-organ damage.[4],[5] The term SCD encompasses the homozygous (hemoglobin SS [HbSS]) and other double heterozygous hemoglobin phenotypes such as hemoglobin S plus C (HbSC), hemoglobin S plus D (HbSD), hemoglobin S plus E (HbSE), and beta thalassemia (HbS/β-thalassemia).[6] The clinical severity of SCD ranges from milder disease phenotypes in HbSC, HbSD, and HbSE diseases to more severe manifestations in HbSS and HS/β-thalassaemia, with poorer disease outcome.[7]
The homozygous HbSS is associated with severe disease presentation, and it is the most common variant in Nigeria and parts of West Africa.[8] Clinically, patients present with recurrent acute manifestations known as “crises” which is interspersed with periods of apparent normalcy, known as steady states.[9] Recurrent acute and chronic pain form part of the clinical spectrum of disease, while the former (known commonly as vaso-occlusive crisis) is typical of the crises of SCD, the latter (including avascular necrosis, commonly of the hip and shoulder joints) represents a long-term disease complication. These have been reported to have a negative impact on the quality of life (QOL) of affected individuals, often associated with depression, disability, unemployment, and opioid analgesics dependence.[10],[11]
SCD is associated with high disease-related mortality arising mainly from complications of severe anemia and end-organ dysfunction. These are particularly marked in Sub-Saharan Africa, where up 90% of affected children have been estimated to die before the age of 5 years, in the absence of intervention.[12],[13] Following the turn of the century, however, significant improvement has been witnessed in the management of SCD patients which has translated into improved survival and longevity, particularly in countries with better health indices. Indeed the introduction of some novel therapeutic interventions such as hydroxycarbamide and stem cell transplantation (SCT) have revolutionized the treatment of individuals with this disorder; SCT has even been associated with cure in some series.[14],[15],[16] With increased survival observed in SCD patients following the introduction of these novel treatment modalities, the presence of long-term disease complications has been brought to the fore; these were hitherto less frequently encountered due to high disease-related mortality. Sickle-related psychopathology (which prominently includes comorbid depression) is among the long-term complications which are increasingly reported; however, due to its rather latent nature, the diagnosis could be missed by the unsuspecting clinician.
| Methodology of Relevant Data Retrieval | | |
The literature search for this review was based on PubMed, PubMed Central, Google Scholar, African Index Medicus, and Scopus database sources; no limitation as to the year of publication was applied. The oldest paper retrieved was published in 1989. The following keywords were used in the search: “sickle cell disease,” “psychological dysfunction,” “psychopathology,” “co-morbid depression,” “quality of life,” “disease severity,” “treatment,” and “clinical outcome.”
The search was restricted to depression occurring in the background of SCD, publications in English language, and was conducted in the month of January 2017.
The studies retrieved dealt mainly on the epidemiology, etiopathogenesis, and treatment of comobid depression in SCD, whereas papers dealing primarily with depression not related to SCD were rejected.
All papers identifi ed were assessed by the authors and subsequently retained or rejected based on the above criteria.
The information from these publications is discussed in this review, with a view to highlighting the magnitude of comorbid depression in SCD as well as to emphasize the need for its early recognition and management by health professionals who care for patients with this disorder.
| Disease Chronicity and Severity and the Development of Comorbid Depression in Sickle Cell Disease | | |
Diseases that run a chronic course have been associated with significant psychological burden arising mainly from frequent hospitalizations, the cost of repeated investigations, and treatment with the attendant drain on family finances [Figure 1].[17],[18] SCD follows a chronic and often “drawn-out” clinical course, characterized by crises, interspersed with periods of steady clinical conditions. The crises periods are times of significant morbidity in patients and are commonly heralded by acute exacerbations in symptoms which could manifest as acute painful (vaso-oclussive crisis), sequestration, aplastic, and the so-called hyperhemolytic crises [Figure 2].[19] These are commonly precipitated by “triggers” which are related to a myriad of causes such as infection, physical and emotional exertion, dehydration, acidosis, and exposure to extremes of temperatures.[20],[21] In the presence of appropriate “triggers,” intravascular red cell sickling occurs leading to deformation of the red cell and change in shape to the characteristic sickle cells. Sickle red cells adhere to one another, to platelets, white cells, and the endothelial lining, resulting in vascular occlusion, ischemia, and pain.[22],[23] The resulting reduction in blood flow from vascular occlusion induces more red cell sickling, thus setting up a vicious cycle of increasing red cell sickling, vaso-occlusion with resultant endothelial, and other organ/system dysfunction.
In the light of the above, it is important to emphasize that the overall burden of SCD, including multiple end-organ dysfunction, changes in physique and psyche, with attendant psychological dysfunction could be traced to the frequency and severity of sickle-related crises. In recognition of this, the goal of management of patients with SCD is, therefore, geared toward shortening the periods of crises and prolonging the steady state periods as much as possible, through diverse interventions.[24],[25] Other sickle-related crises, such as aplastic, hyperhemolytic, and organ sequestration characteristically cause anemia which could lead to hospitalization and blood transfusion, to correct symptomatic anemia [Figure 2]. Recurrent anemia (with acute exacerbations which could occur in sickle-related hyper-hemolysis) is associated with poorer QOL and clinical outcome and has equally been shown to underlie a number of sickle-related complications such as skeletal abnormalities and organ failure [Figure 2].[26] On account of this, it is thus important that anemia is recognized in SCD patients and decision as per red cell transfusion taken early to reduce morbidity and mortality. Even though red cell transfusion (particularly exchange blood transfusions) could signifi cantly improve anemia, reduce complications. and impact positively on wellbeing, complications ranging from alloimmunization, transfusion-related acute lung injury, increased risk of acquiring transfusion transmissible infections (TTIs), and iron overload could occur.[27],[28],[29],[30],[31] Iron overload is a recognized cause of significant morbidity in SCD patients who have transfusion-dependent anemia, arising from iron deposition and organ damage, especially cardiac, hepatic, and endocrine.[32],[33],[34] Failure of any of these organs could significantly impact on the general well-being and QOL and could be a prelude to the development or exacerbation of a frank depressive illness or other psychopathology in SCD participants [Figure 2]. The general prevalence of TTIs in SCD patient tends to parallel the degree of application of safe blood transfusion practices and increases the overall disease burden, particularly in resource-poor settings. Studies in parts of Nigeria and Togo have confirmed that SCD patients on regular blood transfusion could be prone to acquiring TTIs.[35],[36] Infection with any of the TTIs (particularly the hepatotropic viruses) could remarkable increase the hepatopathy of SCD, with a significant increase in disease burden and risk of depression. It is thus evident from the foregoing that the chronic and often debilitating nature of the SCD, which has recurrent acute and chronic pain, increased risk of organ dysfunction, drug dependence, and family and financial stressors as some of its hallmarks, clearly makes a perfect “breeding ground” for the development of comorbid depressive disorder.
| The Epidemiology of Comorbid Depression in Subjects With Sickle Cell Disease | | |
Comorbid depression in subjects living with SCD is frequently not identified or even misdiagnosed by health-care professionals that are routinely charged with the responsibility of managing individuals with this disorder.[37] In this unrecognized state, comorbid depression may only become apparent when significant loss of routine ability and function have occurred in these subjects, a situation that significantly increases both the cost of care and the time to full recovery.[38]
The reported rates of co-depression in SCD subjects are generally comparable to that reported for other chronic medical disorders and ranges from 18% to 44% but are significantly increased when compared to the general population prevalence (even after controlling for illness-related physical symptoms).[10],[39],[40] Previous study among Nigerian subjects with SCD showed a higher prevalence rate of depression than in patients with cancer or malaria but lower than those with HIV/AIDS.[41] The prevalence of comorbid depression has equally been found to be higher in children and adolescents, mainly because children experience high rates of fatigue and other somatic complaints, tend to have more impaired self-esteem and feelings of hopelessness, occasioned by frequent hospitalizations, absences from school, and the inability to experience a normal childhood.[42]
A number of other studies have equally shown that African-American adults experienced higher prevalence rate of SCD-related depressive illness than do the general population (26% vs. 9.5%, respectively).[43],[44] Similarly, the higher prevalence rate of other forms of psychosocial dysfunction in adolescent population with SCD including problems in social relationships, isolation, and school failure have been reported; 29% of participants with these satisfied the criteria for the diagnosis of depression, using the symptom scales.[45] In a study of 440 adult SCD patients, 43% showed depressive symptoms, whereas in another study, 63% (of 38 participants) showed positive psychiatric morbidity, comprising mixed anxiety and depressive symptoms.[46],[47]
Jerrell et al. reported depression prevalence of 46% in SCD patients (90% of which had dysthymia, whereas 10% was diagnosed with major depression) and was associated with adverse course and outcomes.[48] The study equally emphasized the need for early and sustained treatment of comorbid depression in patients with SCD, either by the primary care providers or psychiatrists with a view to reducing the chronic, severe pain-depression cycle.[48]
| The Depression-Pain Cycle | | |
Over the years, the goal of management of diseases that follow a chronic clinical pattern had involved appropriate and adequate symptom management in such a way as to maintain an acceptable QOL.[49],[50] The QOL is very important as it is related to the general outlook of the patient which in turns hinges on the desire to live (to survive). The desire to live is needed in patients with chronic diseases as it guarantees patients' cooperation and active participation in decision-making and treatment.
Sickle-related acute and chronic pains are important symptom complexes of variable duration and intensity experienced by patients and have been shown to have significant adverse impact on QOL and mortality.[51] It has been documented that depression not only commonly complicates chronic pain but also could significantly lower the threshold for tolerance of pain as well as the ability to cope with it.[52] Persistent chronic pain in individuals with SCD may equally induce withdrawal from interpersonal contacts and consequent self-absorption.[52] A withdrawn, self-absorbed patient is more likely to show negative health-seeking behaviors and other activities that could impact adversely on clinical disease severity such as abstinence from follow-up clinic visits as well as discontinuation of the use of prescribed routine medications. In addition, the SCD patient with recurrent pain may exhibit anger and resentment and increased predisposition to opiate use and dependence, particularly when treatment is perceived to be delayed or denied.[52] An earlier study emphasized this observation by reporting that pain severity was correlated with depressed mood, hopelessness, anger, and shame in SCD patients who experience frequent bone pain crises.[53] Ironically, dependence on opiate and other analgesic drug has been shown to induce depression, especially following prolonged usage [Figure 1].[52]
The acute painful crisis of SCD has been attributed to be a common cause of frequent emergency room visits and hospitalizations, absence from school and work, loss of revenue, and increase in other disease-associated stressors to both patient and family.[54],[55],[56] The influence of sickle bone pain crises on disease morbidity has been widely emphasized in a number of reports where these have been consistently linked with adverse prognosis and poor outcome.[57]
Correspondingly, an American study had reiterated that frequent pain crises were significantly associated with depression, somatization, and more frequent hospitalizations.[58] Against the backdrop of this “unholy alliance” between sickle-related pain and reduced physical functioning and subsequently comorbid depression, it is very important that the cycle of pain is truncated in SCD patients to reduce its adverse impact on QOL. This is even made more imperative by the observation that poorly managed acute and chronic pains of SCD could lead to poor coping strategies, which could predispose to higher frequency of vaso-occlusive pain, opioid analgesic dependence, increased emergency room visits and hospital admissions, increased sickle-related complications, poorer physical function, and further reduction in the QOL.[59],[60] The management of sickle-related pains should include interventions for acute pain as well as management aimed at addressing chronic pain and its sequelae. Aside from specific interventions which include different forms of analgesia, other modalities for addressing pain in SCD include the use of complementary and alternative medicine such as prayers to improve coping and well-being.[61],[62],[63] Indeed an increasing number of studies in SCD populations have revealed that prayer and a belief in God are important coping strategies that could be applied in patients with significant benefits.[64],[65],[66],[67]
Due to a combination of chronic anemia, frequent stroke (including silent infarcts), and recurrent bone pain crises, SCD patients tend to have delayed growth and development, which typically manifests as the so-called SCD facie (lower body weight, bony deformities, and an asthenic stature), cognitive impairment/delays, delayed puberty, and missed school days.[68],[69] These complications get reenforced by the period of adolescence and could present as feelings of inadequacy, lower self-esteem, social isolation, poor school performance, and general hopelessness.[70],[71] In extreme cases, suicidal thoughts have been observed; indeed rates of successful suicides are reported to be higher in SCD patients, compared to other patients with chronic diseases.[72]
| Possible Lines of Pharmacological Interventions for Comorbid Depression in Sickle Cell Disease | | |
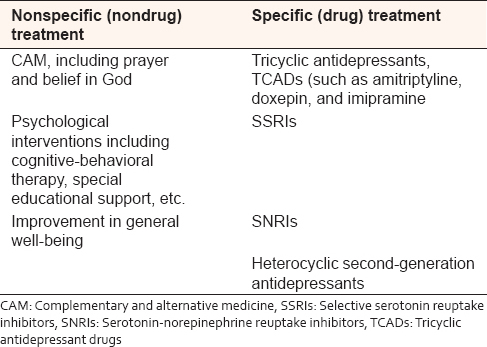
In general, psychological dysfunction in patients with SCD, including major depression, has been successfully treated with a number of modalities which include the use of antidepressants and other interventions aimed at improving the QOL and ameliorating disease course.[10],[73] It will, however, appear that there is suboptimal treatment uptake for SCD patients with depression, arising mainly from low recognition (low index of suspicion) by physicians.[73] Barakat et al. reported that only one-third of patients with SCD reported receiving treatment for depression from a mental health professional.[62] Traditionally, affected individuals are commonly offered any of the tricyclic antidepressants (such as amitriptyline, doxepin, and imipramine) [Table 1]. However, with the turn of the century, newer antidepressants (such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and heterocyclic second-generation agents) are becoming more popular among clinicians [Table 1].[63] These new generation antidepressants have been observed to be safer and more user-friendly, with attendant better compliance, compared to the traditional agents.[63] Drawing from the observation that combining antidepressants with opioid analgesics is significantly associated with fewer hospital visits as a result of sickle-related vaso-occlusive pain,[74] careful combination of these two drugs may actually be indicated in a properly selected group of patients. The use of antidepressants for comorbid depression may be associated with drug-related adverse effects such as bleeding and bone marrow suppression.[43] It is important that physicians and caregivers are mindful of the possibility of these adverse effects since they could become exaggerated in SCD patients that are concomitantly on nonsteroidal anti-inflammatory drugs and hydroxycarbamide, respectively.
It is equally very important to observe that drug treatment alone does not completely address the spectrum of depressive disorders in these patients; therefore, psychological interventions including patient education, cognitive-behavioral therapy, and special educational support have been found to significantly improve the QOL and should be given in combination with drug therapy [Table 1].[75] In practice, however, it will appear that these important aspects of therapy for SCD are usually ignored by caregivers and therefore not readily made available to patients that need it, leading to poor performance status and clinical outcome.[62]
| Conclusion | | |
Comorbid depression appears to be largely unrecognized, undertreated, and underreported; this situation is likely to exaggerate in parts of Sub-Saharan Africa, with its health infrastructural deficits and dearth of qualified specialist health-care providers. The above state of affairs may, therefore, constitute a “perpetuating factor” for recurrence of chronic, debilitating bone crises in patients with its attendant effect in engendering a worse disease course, poor school performance, low self-esteem, and overall poorer QOL and individual productivity. Fortunately, it has been widely reported that the use of antidepressants for adjuvant pain relief in SCD is associated with remarkable improvement in comorbid depression, arising from recurrent sickle-related bone pains.
The above analysis has emphasized the need to increase the awareness, assessment, and surveillance of depressive symptoms by all health-care providers involved in the management of patients living with SCD. Early recognition and optimal treatment of comorbid depression are expected to significantly impact on clinical outcome and overall survival in individuals affected by this disorder, particularly those with more severe clinical phenotype.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| References | | |
| 1. | Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: A HuGE review. Am J Epidemiol 2000;151:839-45. 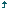 [ PUBMED] |
| 2. | Aneke JC, Okocha CE. Sickle cell disease genetic counseling and testing: A review. Arch Med Health Sci 2016;4:50-7. [Full text] |
| 3. | Okocha C, Onubogu CU, Aneke J, Onah C, Ajuba I, Ibeh N, et al. Prevalence of sickle cell gene among apparently healthy under-two South-East Nigerian children: What is the role of parental premarital counseling and socio-demographic characteristics? Niger J Med 2016;25:176-81. |
| 4. | Frenette PS. Sickle cell vasoocclusion: Heterotypic, multicellular aggregations driven by leukocyte adhesion. Microcirculation 2004;11:167-77. [ PUBMED] |
| 5. | Aneke JC, Adegoke AO, Oyekunle AA, Osho PO, Sanusi AA, Okocha EC, et al. Degrees of kidney disease in Nigerian adults with sickle-cell disease. Med Princ Pract 2014;23:271-4. [ PUBMED] |
| 6. | Lal A, Vichinsky EP. Sickle cell disease. In: Hoffbrand AV, Catovsky D, Tuddenhan EG, editors. Postgraduate Haematology. 5 th ed. Oxford: Blackwell Publishing; 2005. p. 104. |
| 7. | Steinberg MH. Predicting clinical severity in sickle cell anaemia. Br J Haematol 2005;129:465-81. [ PUBMED] |
| 8. | Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: A neglected cause of early childhood mortality. Am J Prev Med 2011;41 6 Suppl 4:S398-405. |
| 9. | Akinbami A, Dosunmu A, Adediran A, Oshinaike O, Phillip A, Vincent O, et al. Steady state hemoglobin concentration and packed cell volume in homozygous sickle cell disease patients in Lagos, Nigeria. Caspian J Intern Med 2012;3:405-9. [ PUBMED] |
| 10. | Hasan SP, Hashmi S, Alhassen M, Lawson W, Castro O. Depression in sickle cell disease. J Natl Med Assoc 2003;95:533-7. [ PUBMED] |
| 11. | Mosaku SK, Oyekunle AA, Aneke JC, Bolarinwa RA, Osho PO, Akinola NO. Avascular necrosis significantly impairs quality of life in sickle cell disease. J Clin Sci 2015;12:41-7. [Full text] |
| 12. | Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: Modelling based on demographics, excess mortality, and interventions. PLoS Med 2013;10:e1001484. [ PUBMED] |
| 13. | Makani J, Cox SE, Soka D, Komba AN, Oruo J, Mwamtemi H, et al. Mortality in sickle cell anemia in Africa: A prospective cohort study in Tanzania. PLoS One 2011;6:e14699. [ PUBMED] |
| 14. | Rees DC. The rationale for using hydroxycarbamide in the treatment of sickle cell disease. Haematologica 2011;96:488-91. [ PUBMED] |
| 15. | Mellouli F, Bejaoui M. The use of hydroxyurea in severe forms of sickle cell disease: Study of 47 Tunisian paediatric cases. Arch Pediatr 2008;15:24-8. [ PUBMED] |
| 16. | Shenoy S. Hematopoietic stem cell transplantation for sickle cell disease: Current practice and emerging trends. Hematology Am Soc Hematol Educ Program 2011;2011:273-9. [ PUBMED] |
| 17. | Watkins KW, Connell CM, Fitzgerald JT, Klem L, Hickey T, Ingersoll-Dayton B. Effect of adults' self-regulation of diabetes on quality-of-life outcomes. Diabetes Care 2000;23:1511-5. [ PUBMED] |
| 18. | Corbin J, Strauss A. Commentary of chronic illness trajectory model. Sch Inq Nurs Pract 1991;5:243-8. |
| 19. | Beyer JE, Simmons LE, Woods GM, Woods PM. A chronology of pain and comfort in children with sickle cell disease. Arch Pediatr Adolesc Med 1999;153:913-20. [ PUBMED] |
| 20. | Tewari S, Brousse V, Piel FB, Menzel S, Rees DC. Environmental determinants of severity in sickle cell disease. Haematologica 2015;100:1108-16. [ PUBMED] |
| 21. | Khan SA, Damanhouri G, Ali A, Khan SA, Khan A, Bakillah A, et al. Precipitating factors and targeted therapies in combating the perils of sickle cell disease - A special nutritional consideration. Nutr Metab (Lond) 2016;13:50. [ PUBMED] |
| 22. | Okpala I. The intriguing contribution of white blood cells to sickle cell disease - A red cell disorder. Blood Rev 2004;18:65-73. [ PUBMED] |
| 23. | Kato GJ, Hebbel RP, Steinberg MH, Gladwin MT. Vasculopathy in sickle cell disease: Biology, pathophysiology, genetics, translational medicine, and new research directions. Am J Hematol 2009;84:618-25. [ PUBMED] |
| 24. | Adewoyin AS. Management of sickle cell disease: A review for physician education in Nigeria (sub-saharan Africa). Anemia 2015;2015:791498. [ PUBMED] |
| 25. | Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, et al. Beyond the definitions of the phenotypic complications of sickle cell disease: An update on management. ScientificWorldJournal 2012;2012:949535. [ PUBMED] |
| 26. | Beverung LM, Strouse JJ, Hulbert ML, Neville K, Liem RI, Inusa B, et al. Health-related quality of life in children with sickle cell anemia: Impact of blood transfusion therapy. Am J Hematol 2015;90:139-43. [ PUBMED] |
| 27. | Chou ST, Liem RI, Thompson AA. Challenges of alloimmunization in patients with haemoglobinopathies. Br J Haematol 2012;159:394-404. [ PUBMED] |
| 28. | Scheunemann LP, Ataga KI. Delayed hemolytic transfusion reaction in sickle cell disease. Am J Med Sci 2010;339:266-9. [ PUBMED] |
| 29. | El Kenz H, Van der Linden P. Transfusion-related acute lung injury. Eur J Anaesthesiol 2014;31:345-50. [ PUBMED] |
| 30. | Bolton-Maggs PH, Cohen H. Serious Hazards of Transfusion (SHOT) haemovigilance and progress is improving transfusion safety. Br J Haematol 2013;163:303-14. [ PUBMED] |
| 31. | Aneke JC, Huntley N, Porter J, Eleftheriou P. Effect of automated red cell exchanges on oxygen saturation on-air, blood parameters and length of hospitalization in sickle cell disease patients with acute chest syndrome. Niger Med J 2016;57:190-3. [ PUBMED] [Full text] |
| 32. | Stanley HM, Friedman DF, Webb J, Kwiatkowski JL. Transfusional iron overload in a cohort of children with sickle cell disease: Impact of magnetic resonance imaging, transfusion method, and chelation. Pediatr Blood Cancer 2016;63:1414-8. |
| 33. | Oduor H, Minniti CP, Brofferio A, Gharib AM, Abd-Elmoniem KZ, Hsieh MM, et al. Severe cardiac iron toxicity in two adults with sickle cell disease. Transfusion 2017;57:700-4. [ PUBMED] |
| 34. | Badawy SM, Liem RI, Rigsby CK, Labotka RJ, DeFreitas RA, Thompson AA. Assessing cardiac and liver iron overload in chronically transfused patients with sickle cell disease. Br J Haematol 2016;175:705-13. |
| 35. | Bolarinwa RA, Aneke JC, Olowookere SA, Salawu L. Seroprevalence of transfusion transmissible viral markers in sickle cell disease patients and healthy controls in Ile-Ife, South-Western Nigeria: A case-control study. J Appl Hematol 2015;6:162-7. [Full text] |
| 36. | Ségbéna AY, Prince-David M, Kagoné TS, Dagnra AY. Human immunodeficiency virus, hepatitis C virus and hepatitis B viruses in patients with sickle-cell disease in Togo. Transfus Clin Biol 2005;12:423-6. |
| 37. | Atkin K, Ahmad WI. Living a 'normal' life: Young people coping with thalassaemia major or sickle cell disorder. Soc Sci Med 2001;53:615-26. [ PUBMED] |
| 38. | Chambliss CR, Heggen J, Copelan DN, Pettignano R. The assessment and management of chronic pain in children. Paediatr Drugs 2002;4:737-46. [ PUBMED] |
| 39. | Laurence B, George D, Woods D. Association between elevated depressive symptoms and clinical disease severity in African-American adults with sickle cell disease. J Natl Med Assoc 2006;98:365-9. [ PUBMED] |
| 40. | Molock SD, Belgrave FZ. Depression and anxiety in patients with sickle cell disease: Conceptual and methodological considerations. J Health Soc Policy 1994;5:39-53. [ PUBMED] |
| 41. | Ehigie BO. Comparative analysis of the psychological consequences of the traumatic experiences of cancer, HIV/AIDS, and sickle cell anemia patients. IFE Psychologia 2003;11:34-54. |
| 42. | Becker M, Axelrod DJ, Oyesanmi O, Markov DD, Kunkel EJ. Hematologic problems in psychosomatic medicine. Psychiatr Clin North Am 2007;30:739-59. [ PUBMED] |
| 43. | Jenerette C, Funk M, Murdaugh C. Sickle cell disease: A stigmatizing condition that may lead to depression. Issues Ment Health Nurs 2005;26:1081-101. [ PUBMED] |
| 44. | Burlew K, Telfair J, Colangelo L, Wright EC. Factors that influence adolescent adaptation to sickle cell disease. J Pediatr Psychol 2000;25:287-99. [ PUBMED] |
| 45. | Benton TD, Ifeagwu JA, Smith-Whitley K. Anxiety and depression in children and adolescents with sickle cell disease. Curr Psychiatry Rep 2007;9:114-21. [ PUBMED] |
| 46. | Wison Schaeffer JJ, Gil KM, Burchinal M, Kramer KD, Nash KB, Orringer E, et al. Depression, disease severity, and sickle cell disease. J Behav Med 1999;22:115-26. [ PUBMED] |
| 47. | Udofia O, Oseikhuemen AE. Psychiatric morbidity in patients with sickle cell anaemia. West Afr J Med 1996;15:196-200. [ PUBMED] |
| 48. | Jerrell JM, Tripathi A, McIntyre RS. Prevalence and treatment of depression in children and adolescents with sickle cell disease: A retrospective cohort study. Prim Care Companion CNS Disord 2011;13. pii: PCC.10m01063. |
| 49. | Barakat LP, Patterson CA, Daniel LC, Dampier C. Quality of life among adolescents with sickle cell disease: Mediation of pain by internalizing symptoms and parenting stress. Health Qual Life Outcomes 2008;6:60. [ PUBMED] |
| 50. | Strickland OL, Jackson G, Gilead M, McGuire DB, Quarles S. Use of focus groups for pain and quality of life assessment in adults with sickle cell disease. J Natl Black Nurses Assoc 2001;12:36-43. [ PUBMED] |
| 51. | Šsberg AN, Heuch I, Hagen K. The mortality associated with chronic widespread musculoskeletal complaints: A systematic review of the literature. Musculoskeletal Care 2017;15:104-113. |
| 52. | Alao AO, Dewan MJ, Jindal S, Effron M. Psychopathology in sickle cell disease. West Afr J Med 2003;22:334-7. [ PUBMED] |
| 53. | Barbarin OA, Whitten CF, Bonds SM. Estimating rates of psychosocial problems in urban and poor children with sickle cell anemia. Health Soc Work 1994;19:112-9. [ PUBMED] |
| 54. | Fletcher C. Appraisal and coping with vaso-occlusive crisis in adolescents with sickle cell disease. Pediatr Nurs 2000;26:319-24. [ PUBMED] |
| 55. | Tsao JC, Jacob E, Seidman LC, Lewis MA, Zeltzer LK. Psychological aspects and hospitalization for pain crises in youth with sickle-cell disease. J Health Psychol 2014;19:407-16. [ PUBMED] |
| 56. | Crosby LE, Joffe NE, Irwin MK, Strong H, Peugh J, Shook L, et al. School performance and disease interference in adolescents with sickle cell disease. Phys Disabil 2015;34:14-30. [ PUBMED] |
| 57. | Brandow AM, Brousseau DC, Panepinto JA. Postdischarge pain, functional limitations and impact on caregivers of children with sickle cell disease treated for painful events. Br J Haematol 2009;144:782-8. [ PUBMED] |
| 58. | Platt OS, Thorington BD, Brambilla DJ, Milner PF, Rosse WF, Vichinsky E, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11-6. [ PUBMED] |
| 59. | Hurtig AL, Park KB. Adjustment and coping in adolescents with sickle cell disease. Ann N Y Acad Sci 1989;565:172-82. [ PUBMED] |
| 60. | Palermo TM, Riley CA, Mitchell BA. Daily functioning and quality of life in children with sickle cell disease pain: Relationship with family and neighborhood socioeconomic distress. J Pain 2008;9:833-40. |
| 61. | Harrison MO, Edwards CL, Koenig HG, Bosworth HB, Decastro L, Wood M. Religiosity/spirituality and pain in patients with sickle cell disease. J Nerv Ment Dis 2005;193:250-7. [ PUBMED] |
| 62. | Barakat LP, Schwartz LA, Simon K, Radcliffe J. Negative thinking as a coping strategy mediator of pain and internalizing symptoms in adolescents with sickle cell disease. J Behav Med 2007;30:199-208. [ PUBMED] |
| 63. | McIntyre RS, Jerrell JM. Polypharmacy in children and adolescents treated for major depressive disorder: A claims database study. J Clin Psychiatry 2009;70:240-6. [ PUBMED] |
| 64. | Thompson WE, Eriator I. Pain control in sickle cell disease patients: Use of complementary and alternative medicine. Pain Med 2014;15:241-6. [ PUBMED] |
| 65. | Cooper-Effa M, Blount W, Kaslow N, Rothenberg R, Eckman J. Role of spirituality in patients with sickle cell disease. J Am Board Fam Pract 2001;14:116-22. [ PUBMED] |
| 66. | Cotton S, Grossoehme D, McGrady ME. Religious coping and the use of prayer in children with sickle cell disease. Pediatr Blood Cancer 2012;58:244-9. [ PUBMED] |
| 67. | Bediako SM, Lattimer L, Haywood C Jr, Ratanawongsa N, Lanzkron S, Beach MC. Religious coping and hospital admissions among adults with sickle cell disease. J Behav Med 2011;34:120-7. [ PUBMED] |
| 68. | Pithon MM, Palmeira LM, Barbosa AA, Pereira R, de Andrade AC, Coqueiro Rda S. Craniofacial features of patients with sickle cell anemia and sickle cell trait. Angle Orthod 2014;84:825-9. [ PUBMED] |
| 69. | Burkhardt L, Lobitz S, Koustenis E, Rueckriegel SM, Hernaiz Driever P. Cognitive and fine motor deficits in a pediatric sickle cell disease cohort of mixed ethnic origin. Ann Hematol 2017;96:199-213. |
| 70. | Lee EJ, Phoenix D, Brown W, Jackson BS. A comparison study of children with sickle cell disease and their non-diseased siblings on hopelessness, depression, and perceived competence. J Adv Nurs 1997;25:79-86. [ PUBMED] |
| 71. | Seigel WM, Golden NH, Gough JW, Lashley MS, Sacker IM. Depression, self-esteem, and life events in adolescents with chronic diseases. J Adolesc Health Care 1990;11:501-4. [ PUBMED] |
| 72. | Edwards CL, Green M, Wellington CC, Muhammad M, Wood M, Feliu M, et al. Depression, suicidal ideation, and attempts in black patients with sickle cell disease. J Natl Med Assoc 2009;101:1090-5. [ PUBMED] |
| 73. | Taylor LE, Stotts NA, Humphreys J, Treadwell MJ, Miaskowski C. A biopsychosocial-spiritual model of chronic pain in adults with sickle cell disease. Pain Manag Nurs 2013;14:287-301. [ PUBMED] |
| 74. | Jerrell JM, Tripathi A, Stallworth JR. Pain management in children and adolescents with sickle cell disease. Am J Hematol 2011;86:82-4. [ PUBMED] |
| 75. | Levenson JL, McClish DK, Dahman BA, Bovbjerg VE, de A Citero V, Penberthy LT, et al. Depression and anxiety in adults with sickle cell disease: The PiSCES project. Psychosom Med 2008;70:192-6. [ PUBMED] |
[Figure 1], [Figure 2]
[Table 1]
|

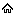







 Search Pubmed for
Search Pubmed for