A New Class of Estrogen Receptor Beta–Selective Activators
Estrogen action is mediated by binding to two specific estrogen receptors (ERs), ERα and ERβ. The discovery of ERβ in 1996 changed our understanding of estrogen action and sparked intense research efforts to discover its role in normal physiology and its potential as a drug target. Several ERβ selective and clinically active ligands have since been developed. The first compounds described were those that have high selective binding affinity for ERβ, such as ERB-041. A second class of agents that have been identified bind to ERβ and ERα with similar affinity but selectively activate ERβ, such as the flavanone liquiritigenin. A recent study has identified, 3,3′-diindolylmethane (DIM) as a new class of ERβ activator that does not bind to the ligand binding site, but rather selectively activates ERβ possibly through cellular kinase pathways that target the receptor’s ligand-independent activation domain. Although more studies are needed, these findings suggest that compounds that modulate of ERβ activation without directly binding to the receptor might prove to be of significant clinical importance in the future.
Estrogens are a family of steroid hormones that play important roles in reproduction, and regulate many biological processes. Their actions are mediated by binding to estrogen receptors (ERs), which are members of the nuclear receptor (NR) superfamily and function as ligand-regulated transcription factors (1). Prior to the mid 1990s, estrogen signaling was thought be mediated by a single ER (now known as ERα; NR3A1); however, the report of a second ER (ERβ; NR3A2) in 1996 (2) caused a dramatic shift in our understanding of estrogen action. Studies of ERα- or ERβ-null mice demonstrated that each subtype has similar but also unique roles in estrogen action. ERα is primarily expressed in the uterus, liver, kidney, and ovarian theca cells, whereas ERβ is predominantly expressed in the prostate, lung, bladder, and granulosa cells. ERα and ERβ are also coexpressed in a number of tissues and can heterodimerize, with the expression of ERβ positively or negatively impacting ERα-dependent transcription under specific conditions (3, 4).
Similar to other members of the NR superfamily, ERs consist of different functional domains (Figure 1). A centrally located zinc-finger DNA-binding domain (DBD) is followed by a flexible hinge region and a C-terminally located ligand binding domain (LBD). ERα and ERβ share a high degree of sequence identity in their DBDs (96%) but not in their LBDs (56%). Nonetheless, both receptors bind to their specific DNA recognition sequence referred to as estrogen responsive element (ERE) and interact with the endogenous estrogen [17β-estradiol (E2)] with similar affinities (2). Ligand binding triggers conformational changes in ERs leading to dimerization, DNA binding, and recruitment of coregula-tor proteins (coactivators or corepressors) to regulate target gene expression (5). The transactivating functions of ERα and ERβ are mediated by two activation domains, referred to as activation function (AF-1) and activation function (AF-2) (5). The AF-2 region, located in the LBD, is responsible for ligand-dependent activation and recruitment of coactivator proteins, including members of the nuclear receptor coactivator family (NCoA) [also known as steroid receptor coactivator family (SRC)] (6). When the LBD of either receptor is bound by an agonist, helix 12 located within the AF-2 domain becomes positioned over the ligand-binding pocket and forms an interaction surface that is recognized by coactivator proteins (7). The ligand-independent N-terminally located AF-1 domains of ERα and ERβ exhibit less than 20% sequence identity. AF-1 function, which is cell type– and promoter-specific, is known to be regulated by growth factor and cellular protein kinases (8, 9). Specific serine residues within the AF-1 domains of ERα and ERβ are target sites for protein kinase signaling cascades, such as those mediated by mitogen-activated protein kinase (MAPK) and phos-phoinositide 3-kinase–Akt (PI3K–Akt). Early reports suggested that ERβ AF-1 function is minimal compared to that of ERα (10); however, phosphorylation of AF-1 serine residues in ERβ enhances ligand-independent interaction with the coactivator NCoA1 (9).
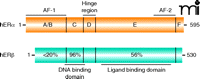
Schematic representation of the structural domains of human estrogen receptor α and β. The percentages represent the amino-acid sequence identity of the different functional domains in ERβ compared to those of ERα.
One of the important findings from the discovery of ERβ was that estrogen signaling is a carefully regulated balancing act between ERα and ERβ (11). ERα promotes cell proliferation whereas ERβ is anti-proliferative (12). Moreover, it became apparent early on that certain estrogenic compounds exhibited modest ER–subtype selectivity. Genistein, a natural isoflavone, was one of the first compounds reported to have selective affinity for ERβ, which is approximately thirty-fold higher than that for ERβ (13). These functional differences and distinct ligand binding preferences led to excitement at the prospect of developing ERβ-selective and clinically relevant compounds. Several ERβ selective ligands have since been developed and many of them are being evaluated in phase II and III clinical trials. The different ERβ selective compounds can be divided into distinct functional classes based on their mechanism of ER subtype selectivity (Table 1). The first class of ligands are based on the exploitation of the ERβ pharmacophore that best positions compounds to interact preferentially with ERβ using structure-based drug design and molecular modeling approaches. One of the first synthetic ERβ selective compounds reported to have greater relative affinity for ERβ compared to genistein was 2,3-bis(4-hydroyphenyl)propionitrile (DPN) (14). DPN acts as an agonist for both ER subtypes, but has a seventyfold higher binding affinity and 170-fold greater ability to activate ERβ in transcription assays (14). Other ERβ-selective ligands have been described by academic laboratories and pharmaceutical companies, with ERB-041 (2-(3-fluro-4-hydroxyphenyl)-7-vinyl-1,3-benzoxazol-5-ol) being most studied (15). ERB-041 has a 200-fold higher binding affinity for ERβ compared to ERα, but does not display a further increase in subtype selectivity in transcription assays (15). ERB-041 is not active in uterus and mammary gland, but has been found to be highly beneficial in pre-clinical rodent models for inflammatory bowel disease, rheumatoid arthritis and endometriosis (15, 16). These findings and work by others support a role of ERβ-selective ligands in the treatment of inflammatory disorders. Other laboratories have focused their efforts on identifying natural compounds with ERβ selectivity. Menopause formula 101 (MF101), a botanical extract generated from twenty-two different plant species, selectively activates ERβ and decreases the frequency of hot flashes in a phase II clinical trial (17). Liquiritigenin, a flavanone present in Glycyrrhizae uralensis, was later isolated as the most active estrogenic component of MF101 (18). Unlike DPN and ERB-041, liquiritigenin binds to both ER subtypes with similar affinity but activates ER target genes solely through ERβ (18). This ERβ-selectivity arises from the specific recruitment of nuclear receptor coactivator 2 (NCoA2) to estrogen-responsive genes. Thus, liquiritigenin represents a second class of ERβ-selective compounds that bind similarly to ERα and ERβ but only activate ERβ. It is note worthy that a tetrahydrochrysene (THC) derivative which has a modest fourfold preferential binding affinity for ERβ is a complete ERβ antagonist but an ERα agonist (19). Whether liquiritigenin antagonizes activated ERα has not been reported (18). The selective activation of ERβ may be influenced by the coactivator composition present in the cells, and, under the right cellular conditions, liquiritigenin might activate ERα. Evaluating the ERβ selectivity in a variety of cell lines and different tissues will be important to fully determine the subtype selectivity of liquiritigenin.
Classification of ERβ-Selective Compounds
A recent paper from Vivar and colleagues identified 3,3′-diindolylmethane (DIM) as a third class of ERβ selective compounds that do not bind to either ERα or ERβ but selectively activate ERβ in a ligand-independent manner (20). DIM is the major in vivo product derived from the acid-catalyzed condensation of indole-3-carbinol (I3C), which is a chemoprotective agent found in Brassica vegetables, including broccoli, cauliflower, and brussels sprouts. Using levels achievable by dietary intake, the authors report that DIM selectively activated ERβ in transient transfection assays, using an ERE-regulated reporter gene. These findings were confirmed in U2OS human osteosarcoma cells that stably express either ERα or ERβ, in which the authors showed that DIM selectively activated multiple endogenous target genes solely through ERβ. The authors used chromatin immunoprecipitation (ChIP) assays to show that DIM induced the selective recruitment of ERβ and NCoA2 to 5′-regulatory region of keratin 19 gene, one of three genes examined. RNAi-mediated knockdown of NCoA2 expression prevented DIM-induced activation of keratin 19, revealing NCoA2 as an important piece to this puzzle. Although the precise mechanism by which DIM activates ERβ is unknown, the authors suggested that ERβ and DIM may be linked through the activation of cellular kinases (e.g., MAPK) indicating a role for the AF-1 domain. However, evaluation of the ability of DIM to activate ERβ in the presence of specific kinase inhibitors was not performed. Studies in ERα-positive MCF-7 human breast cancer cells showed that DIM induced ERα to bind to an ERE motif and to activate transcription of E2-responsive genes (21). DIM was found to be a potent ligand-independent activator of ERα through protein kinase A and MAPK pathways in Ishikawa human endometrial adenocarcinoma cell lines (22). Other investigators have also reported DIM-dependent recruitment of ERα to estrogen responsive genes (23, 24). Therefore, it will be important to confirm the ERβ-selectivity of DIM using a panel of cell lines that express endogenous ERβ as well as the appropriate animal models.
DIM is known to activate multiple signaling pathways, including those that involve nuclear factor-kappaB (NF-κB) signaling, caspase activation, DNA repair, cytochrome P450 metabolism, as well as various cellular kinases (25). DIM also binds and activates the aryl hydrocarbon receptor (AHR) (26, 27). Recently, the selective estrogen receptor modulator 4-hydroxytamoxifen was found to mediate its suppressive effects on osteoclast differentiation, in part through the AHR (28). The AHR nuclear translocator (ARNT), which is the obligatory heterodimerization partner of AHR [for example, see (29, 30)], also is a potent AF-1-dependent coactivator of ERβ (31). It will be interesting to determine the role, if any, of AHR signaling in the ERβ-dependent action of DIM.
Human ERβ is translated from exons 1–8 to encode the 530-residue full-length receptor. Several ERβ isoforms have been identified that arise from alternative splicing of exon 8 and exon 9 (11, 32). ERβcx (also called ERβ2) is one of the best-studied variants and is identical to ERβ except that twenty-six unique aminoacid residues from exon 9 replace exon 8 (32). ERβcx does not bind ligand, lacks a functional AF-2 region, and shows preferential heterodimerization with ERα rather than with ERβ, thereby inhibiting ERα DNA binding and having a dominant-negative effect on ligand-dependent ERα reporter gene activity. It is tempting to speculate that DIM might also activate this and perhaps other ERβ variants.
Although the results reported by Vivar et al. are exciting, further characterization of DIM is needed to fully evaluate the ERβ selectivity of this compound (20). Their findings also remind us of the importance of ligand-independent activation in ER signaling, which is an aspect of ER action that still remains poorly understood. Studies of modified DIM derivatives reveal that this compound is a very good starting structure for the development of new anti-cancer agents and unique receptor agonists. DIM analogs containing a p-trifluoromethylphenyl, p-t-butylphenyl, or p-biphenyl group activate peroxisome proliferator-activated receptor γ (PPARγ) whereas p-methoxyphenyl- or p-phenyl–containing DIM compounds activate nerve growth factor-induced receptor Bα (NGFI-Bα also known as Nur77) [reviewed in (33)]. One of the many challenges will be to ensure that the in vitro effects observed by DIM are specific to ERβ since DIM activates many other signaling pathways. Therefore, determining the precise molecular targets of DIM that are involved in the activation of ERβ will be important first steps to designing synthetic agents with improved specificity for ERβ. Nonetheless, the findings of the Vivar et al. (20) study open new research opportunities to identify ER selective agents and suggest that compounds that modulate ERβ activation without directly binding to the receptor might prove to be of significant clinical importance in the future.
Acknowledgments
The authors express their appreciation to Shaimaa Ahmed and Laura MacPherson for their helpful suggestions. Research in the laboratory of JM is supported by the Canadian Institute of Health Research, Canadian Breast Cancer Research Alliance and the Canadian Breast Cancer Research Foundation. JM is a recipient of a Canadian Institute of Health Research New Investigator Award.
- Copyright © 2010
References

Raymond Lo, BS, is a PhD student in Dr. Matthews’s laboratory. His research project aims to determine the role that transcription factors play in estrogen- and benzo[a]pyrene-induced oxidative DNA damage and DNA adduct formation in breast cancer models.

Jason Matthews, PhD, is an Assistant Professor in the Department of Pharmacology and Toxicology at the University of Toronto in Canada. The research interests of his laboratory include study aryl hydrocarbon receptor signaling and its modulation of estrogen receptor action and its role in breast cancer. His laboratory also studies the role of estrogen receptors in estrogen-induced oxidative DNA damage. E-mail Jason.matthews{at}utoronto.ca; fax 416-978-6395.

