SOCS3 in Liver Regeneration and Hepatocarcinoma
Cytokines are soluble mediators that regulate numerous cellular process including haematopoiesis, differentiation, survival and immunological functions. Many cytokines signal through the Janus kinase–Signal transducers and activators of transcription (JAK-STAT) pathway to activate transcription of target genes involved in these processes (1). Prolonged cytokine responses (i.e., enhanced expression of cytokines and their receptors, and sustained activation of these receptors) are detrimental and can lead to inflammation, autoimmune diseases, and cancer; thus, cytokine signaling must be kept in check through several different regulatory mechanisms.
The Suppressors of Cytokine Signaling (SOCS) constitute one important family of negative regulators that mediate immune responses (2). Most SOCS proteins are expressed following STAT activation and, therefore, function as classical negative regulators to inhibit cytokine-mediated signal transduction. At present, the SOCS family consists of eight proteins: cytokine-inducible Src homology 2 (SH2) domain–containing protein (CIS) and SOCS1–7. All contain a central SH2 domain and a well-conserved SOCS box located near the C terminus (3). SOCS are believed to inhibit cytokine signaling by either SH2-mediated binding to specific phosphotyrosine residues on activated cytokine receptors and/or JAKs, or by targeting receptor complexes for degradation (4). The latter is achieved by forming ECS (Elongin-Cullin-SOCS box)-containing E3 ubiquitin ligases that accelerate the ubiquitination and degradation of SOCS binding partners (5). This viewpoint focuses on our current understanding of the divergent roles of SOCS3 and discusses some recent findings by Riehle et al. (6), highlighting the importance of SOCS3 in modulating cytokine and neoplastic proliferative responses in the liver.
SOCS1 and SOCS3 are the best-characterized members of the SOCS family; however, their mechanisms of action differ. Unlike SOCS1, SOCS3 only inhibits JAK-STAT signaling when bound to phosphorylated receptors, such as the gp130 receptor at phosphotyrosine 759 (pY759) (7). Mice deficient in SOCS3 die in utero as a consequence of uncontrolled leukemia inhibitory factor (LIF) signaling—inactivation of the LIF receptor rescues the socs3−/− phenotype (8, 9). Conditional socs3 knockouts have been generated that overcome the problems associated with socs3 germline deletions. Myeloid cell–specific SOCS3-deficient mice display sustained Interleukin (IL)-6-mediated activation of STAT3, establishing SOCS3 as a crucial inhibitor of IL-6 signaling in vivo (10). Not surprisingly, SOCS3 deficiency contributes to the progression of chronic inflammatory diseases, such as arthritis, Crohn’s and inflammatory bowel disease (11). Mice lacking SOCS3 in hematopoietic cells appear normal but at approximately seventeen weeks of age develop inflammatory pathologies. When these mice are injected with granulocyte colony-stimulating factor (G-CSF), they develop neutrophilia and splenomegaly, suggesting SOCS3 is a crucial negative regulator of G-CSF signaling (12). Similarly, SOCS3 regulates lysosomal routing of the G-CSF receptor (13).
SOCS3 is also thought to participate in T-cell differentiation. SOCS3 inhibits IL-12-mediated STAT4 activation, thereby blocking T Helper (Th) cell type 1 development (14). Consequently, SOCS3 expression may skew T-cell differentiation toward a Th2 phenotype. Because atopic dermatitis, allergic rhinitis, and bronchial asthma are allergic immune disorders characterized by a predominance of Th2 cells, SOCS3 expression may correlate with the severity of such Th2-related allergies and could become a new therapeutic target for the development of anti-allergic drugs, but this would likely require refined cell-type- and tissue-specific targeting. SOCS3 is also constitutively present up to twenty-four hours following treatment of primary macrophages with lipopolysaccharide (LPS, which is known to cause an inflammatory/immune response), suggesting SOCS3 may promote inflammation under certain circumstances (15). We have proposed that the proinflammatory nature of SOCS3 may be a consequence of its ability to promote the degradation of inflammatory response–inhibiting receptors such as CD33 (16). Because CD33 is a marker of acute myeloid leukemia (AML), these findings may be important strategically regarding CD33-related therapies.
The liver, the central organ for xenobiotic metabolism, possesses strong regenerative capability following surgical resection or toxic liver injury. Specifically, damaged liver tissue is not replaced but the remaining healthy liver proliferates to compensate for lost tissue. IL-6 abets the acute-phase response induced by hepatic inflammation by priming hepatocytes to respond to growth factors [such as epidermal growth factor (EGF)], enabling cell cycle progression and proliferation (17). Signaling of IL-6 through tumor necrosis factor receptor 1 (TNFR1), nuclear factor–κB (NFκB), and STAT3 promotes cell survival and hepatocyte replication but also leads to Socs3 transcription. Thus, SOCS3 expression functions in a negative feedback loop to attenuate IL-6 signaling (Figure 1⇓).
Recent research by Riehle et al. investigates whether SOCS3 might act as a negative regulator of liver regeneration and protect against prolonged cytokine responses. To overcome the problems associated with SOCS3 germline deletions, the authors generated hepatocyte-specific Socs3 knockout mice (h-KO) and analyzed the response of these mice to two-thirds “partial” hepatectomy (PH) compared to the same PH performed on wild-type littermates. In the absence of SOCS3, sustained IL-6 signaling did not lead to cytokine-induced toxicity, nor did it lead to a decrease in cell proliferation. Instead, hepatocyte DNA replication and cell cycle progression were enhanced in Socs3 h-KO mice after PH, leading to accelerated liver regeneration. The authors demonstrated that prolonged activation of STAT3 and extracellular-regulated kinases 1 and 2 [ERK1/2, members of the mitogen-activated protein kinase (MAPK) family] may be responsible for the enhanced proliferative capacity of SOCS3-deficient hepatocytes both in vivo and in vitro. Interestingly, in SOCS3-deficient macrophages, IL-6 appears to induce IFN-γ-like immunosuppression through prolonged STAT1 activation (10). However, in Socs3 h-KO mice, the authors state (referring to unpublished data) that prolonged activation of STAT1 or STAT5 was not observed. It will be interesting and useful if the authors investigate the effects of enhanced signaling of LIF in the liver in the absence of SOCS3. Using an Affymetrix microarray, they concluded that the expression of mRNA for signaling intermediates in the toll-like receptor (TLR), focal adhesion kinase (FAK), and Wnt signaling pathways are similarly increased in Socs3 h-KO mice. These findings suggest that SOCS3 is not only essential in controlling the expression of acute phase responses in the liver but also is an important regulator of hepatocyte proliferation. It is hoped that, by understanding the mechanisms of liver regeneration, effective new therapies may emerge to manage or treat a number of important conditions, including acute liver failure, cirrhosis, and fatty liver degeneration.
The roles of SOCS proteins might grow to include that of tumor suppressors. A plethora of studies have revealed that silencing of Socs gene expression occurs in numerous tumors. In relation to SOCS3, epigenetic gene silencing of SOCS3 by CpG island methylation has been identified in approximately 90% of head-and-neck squamous cell carcinoma samples, in hepatocellular carcimona (HCC), and in non-small-cell lung carcinomas (18–20). Becuase Riehle et al. found enhanced hepatocyte proliferation in Socs3 specific h-KO mice, the authors investigated whether these mice were more prone to developing liver carcinomas. Socs3 h-KO mice or wild-type littermates were injected with a single dose of DEN (N-nitrosodiethtylamine) to induce hepatocarcinogenesis. At nine months of age, 4/6 Socs3 h-KO mice developed HCC, compared with 0/6 wild-type littermates. The cumulative evidence suggests SOCS3 is not only a suppressor of cytokine signaling but also a tumor suppressor, where loss of SOCS3 expression may confer a cell growth advantage and promote cell migration owing to enhanced JAK-STAT and FAK signaling, respectively. Various other reports suggest SOCS3 is stabilized in some tumors. Constitutive SOCS3 expression was observed in situ in infiltrating breast ductal carcinomas (21), and we have proposed that altered SOCS3 function may contribute to myeloproliferative disorders (MPDs) resulting from constitutive activating mutations in JAK2. In MPDs, SOCS3 protein is stabilized; however, we hypothesize that aberrant SOCS3 tyrosine phosphorylation renders SOCS3 functionally inactive by blocking its E3-ligase activity, thus rendering SOCS3 incapable of regulating myelopoiesis (22). Similarly, tyrosine phosphorylated SOCS3 interacts with and prevents the function of the Ras inhibitor p120 RasGAP, thereby maintaining activation of ERK, suggesting that tyrosine phosphorylation of SOCS3 may ensure cell survival (23). Therefore, loss of SOCS3 expression or rendering SOCS3 functionally inactive may have the same outcome whereby SOCS3 is unable to regulate signaling.
Although loss of SOCS3 expression encourages hepatocyte proliferation and survival, questions remain as to whether these discoveries could be applied therapeutically to facilitate liver regeneration or to treat liver carcinomas. Could a SOCS3 small-interfering RNA (siRNA) or a dominant-negative form of SOCS3 be used to promote hepatocyte proliferation? Alternatively, could overexpression of SOCS3 be used to treat hepatocellular carcinoma? It is important to remember liver regeneration means that lost tissue will not be reconstituted but the remaining tissue replicates to replace the lost tissue mass. Studies indicate that a two-thirds PH affords optimal liver regeneration (24), but the effects of SOCS3 deficiency under other hepatectomy procedures need to be addressed. Other negative regulators of liver regeneration, such as Caveolin-1, have been compared functionally to SOCS proteins both as regulators of JAK-STAT signaling and as tumor suppressors (25); these other regulators could also serve as attractive therapeutic alternatives, singly or in combination with SOCS3 targeting. Successful liver regeneration relies heavily on the balance of growth-promoting and growth-inhibitory pathways. It is hoped that further understanding of the pathways leading to liver regeneration and how these pathways are regulated may be successfully exploited to control liver regeneration and tumor growth.
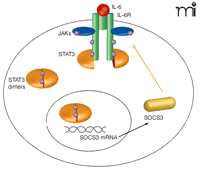
Regulation of IL-6 signaling by SOCS3. IL-6 signaling leads to activation of gp130 and phosphorylation (represented by purple circles) of STAT3 monomers by Janus kinases (JAKs). STAT3 then homodimerizes and translocates to the nucleus, where it induces transcription of a number of target genes, including Socs3. SOCS are believed to inhibit cytokine signaling by either binding to activated cytokine receptors and/or JAKs, or by targeting receptor complexes for degradation. SOCS3 can bind to JAK and the gp130 receptor at phosphotyrosine 759 (pY759) via its kinase inhibitory region (KIR) and Src homology 2 (SH2) domains, respectively, to inhibit JAK-STAT signaling.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Joanne Elliott, PhD, is a Research Fellow at QUB working with Prof. Jim Johnston. Their research focuses on the regulation of SOCS E3 ligase activity and the relevance of SOCS3 phosphorylation in myeloproliferative disease. I would like to thank Jim Johnston for reviewing this manuscript. E-mail joanne.elliott{at}qub.ac.uk; fax + 44 2890325839.

