|
|
Pages du praticien
Un buveur de 74 ans avec perte de mémoire et neuropathie
John C. M. Brust, MD, Discussant
JAMA. 2008;299(9):1046-1054
RÉSUMÉ
| |
Les effets indésirables de l’alcool sur le système nerveux central et périphérique peuvent être directs (neurotoxicité) ou indirects (carence nutritionnelle notamment). En utilisant le cas de Monsieur E., un homme d’âge avancé avec une consommation d’alcool modérée à forte présentant des troubles mnésiques, les orientations diagnostiques, incluant troubles cognitifs légers, maladie d’Alzheimer précoce, syndrome de Wernicke-Korsakoff, et « démence alcoolique », sont discutées. Ces troubles ne sont pas mutuellement exclusifs, et chez un patient souffrant de déficit cognitif léger ou de démence, le rôle contributif de l’alcool peut être difficile à déterminer. En effet, des études épidémiologiques suggèrent que la consommation faible à modérée d’alcool réduit effectivement le risque de développer des troubles cognitifs légers ou une démence, notamment la maladie d’Alzheimer. La prise en charge inclut des mesures visant à réduire l’alcoolodépendance (thérapie comportementale ou traitement pharmacologique par exemple) et à retarder la progression du déficit cognitif (notamment en adoptant des comportements sains comme la pratique de loisirs cognitifs).
DR DELBANCO : Monsieur E. est un buveur de 74 ans, préoccupé par la survenue de troubles de mémoire et de symptômes névropathiques. Avocat à la retraite, il bénéficie du Medicare et d’une assurance privée. Il est célibataire et vit séparé de sa famille. Monsieur E. n’a jamais fumé et pratique régulièrement de l’exercice sur vélo et tapis roulant. Il avait l’habitude de jouer au tennis mais ne s’estime plus vraiment apte à ce sport. Il n’a pas d’antécédents familiaux d’alcoolodépendance ni de maladie neuromusculaire.
Il y a environ 20 ans, Monsieur E. a remarqué un amaigrissement de ses mains et des problèmes de motricité fine. Depuis 10 ans, il ne peut plus ouvrir les bocaux et ses mains sont déformées, avec des difformités bilatérales de ses 2 derniers doigts. Il présente également des orteils en marteau et des « sensations étranges » à la marche dans la plante des pieds.
En 1998, l'examen par un neurologue a révélé une importante atrophie des muscles intrinsèques de la main et conduit à une suspicion de compression bilatérale du nerf ulnaire. Le médecin consultant a également noté un amaigrissement des jambes en dessous du genou. La chirurgie de libération du nerf ulnaire a stabilisé certaines des sensations de faiblesse et des paresthésies dans ses mains, mais n'a pas permis le retour à la normale de leur motricité. Après une nouvelle consultation, parallèlement à l’obtention d’un électromyogramme, les médecins ont pensé que M. E. souffrait d’une neuropathie démyélinisante lentement progressive, vraisemblablement héréditaire, représentant probablement une variante de la maladie de Charcot-Marie-Tooth (CMT).
Pendant une grande partie de sa vie, M. E. a bu « quotidiennement 2 ou 3 verres d’alcool fort » et jusqu’à « 3 à 4 vodkas par jour, parfois plus. » Environ 10 ans plus tôt, il a brusquement arrêté de travailler et s'est mis à boire davantage. Rétrospectivement, il pense que l’augmentation de sa consommation a été essentiellement induite par le vide qu'il a éprouvé en arrêtant de travailler et par son manque d’activité à cette période. Peu de temps après, il s'est mis à « broyer du noir » et a commencé à boire excessivement. Il prenait du « véritable alcool, pas du vin ou de la bière. »
Au cours des 10 dernières années, il a remarqué certaines pertes de mémoire. Il a de plus en plus de mal à se souvenir des noms. Parfois il n’arrive pas à trouver ses mots, et il pense ne plus réagir aussi vite qu’auparavant. Il affirme n’avoir ni étourdissements ni hallucinations.
Il a également développé une hypertension parfois difficile à contrôler. En dehors de 3 herniorraphies, il a toujours été globalement en bonne santé. Actuellement, sa consommation d’alcool est réduite, mais il prend toujours 1 ou 2 verres par jour. Il est traité par vérapamil, hydrochlorothiazide, et lisinopril pour son hypertension, et par ranitidine pour un reflux gastro-oesophagien.
Lors d'une récente consultation chez un neurologue, son affect était intact en dépit d’une élocution assez lente. Son état mental n’a suscité aucun autre commentaire. Il apparaissait en bonne santé. Sa pression artérielle était de 130/80 mm Hg, et elle était également normale chez lui. Son pouls était de 74/min, régulier, et il n'était pas tachypnéique. Il présentait une perte d'audition modérée, et l’une de ses membranes tympaniques portait des traces de cicatrice. L'examen physique général était normal ; rien ne suggérait de maladie hépatique ou de modifications cutanées liées à l'abus d'alcool.
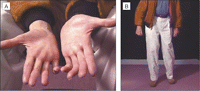
Lors d’évaluations neurologiques récentes, les nerfs crâniens du patient apparaissaient normaux, et il ne présentait aucun tremblement. Les répétitions de l’épreuve doigt-nez étaient normales. Une atrophie marquée des muscles intrinsèques de ses mains était observée, ainsi que des difformités en griffe des quatrième et cinquième doigts, bilatéralement (FIGURE ; voir également la vidéo sur http://jama.ama-assn.org/cgi/content/full/299/9/1046/DC1). Il présentait une atrophie musculaire modérée et une perception réduite des vibrations, des piqûres d’épingle, et de la température en dessous des genoux. Les réflexes tendineux étaient absents dans les membres inférieurs. Des fasciculations ont été observées une fois, mais pas de manière persistante, et ses orteils étaient en griffe. M. E. était incapable de maintenir une marche en tandem et présentait un élargissement du polygone de sustentation (Figure ; voir également la vidéo sur http://jama.ama-assn.org/cgi/content/full/299/9/1046/DC1).
|
|
|
|
Figure. Observations physiques à l’examen des mains et de la démarche de Mr E
A, Examen des mains de Mr E montrant une atrophie des muscles intrinsèques et des deformities en griffe des 4ème et 5ème doigt. B, Mr E était incapable de maintenir une marche coordonnée. (Voir vidéo à http://jama.ama-assn.org/cgi/content/full/299/9/1046/DC1.)
|
|
|
Ses examens biologiques ont révélé une numération formule sanguine normale, avec une numération plaquettaire et un rapport normalisé international normaux. Dans le passé, son volume globulaire moyen (VGM) avait parfois été élevé. Récemment, ses taux de vitamine B12 étaient normaux, bien que situés à la limite inférieure lors de la première mesure effectuée des années plus tôt ; il a en outre reçu des injections de vitamine B12 pendant un temps. Les résultats des tests de la fonction hépatique étaient normaux. Il présentait des taux constamment élevés de créatine-phosphokinase. Ses taux de thyrotropine, d’immunoglobuline, et de folate étaient normaux, et son bilan sanguin était négatif pour le virus de l’immunodéficience humaine.
MONSIEUR E. : SON POINT DE VUE
J'ai travaillé pour une importante multinationale qui avait une politique très stricte en matière d'alcool. Il n'y avait pas d’alcool au bureau. Les soirées en groupe faisaient en fait partie des rares occasions où l’on consommait du vin ou de l'alcool aux frais de la société. Je n'ai pas plongé dans la boisson le jour où j’ai quitté l’entreprise. Le vrai problème a résidé dans le fait que j'ai arrêté assez brutalement de travailler, et que je n’avais jamais vraiment pensé à ce que j’allais faire après. Alors il y a eu un vide dans mon emploi du temps et dans mes pensées. Et je pense que c’est là que j’ai commencé.
J'ai commencé par boire davantage le soir. Au bout d’un moment, je me suis mis à boire à midi. Je ne saurais vraiment pas dire combien je bois. Je n’ai jamais été ivre mort. Je ne suis jamais tombé. Je m’endors parfois après dîner, en regardant la télé par exemple. Le lendemain matin je n'ai pas mal à la tête, je ne suis pas malade, mais je me sens plus apathique que si je n'avais pas trop bu. Les gens me parlent de ce problème. Surtout ma fille qui est adulte ; elle pense que je devrais moins boire.
À certains égards peut-être, ma mobilité n’est plus la même, mais cela ne s’est pas fait en un jour. Cela fait partie de ces choses qui se font lentement et à long terme. Mon médecin et moi parlons de mes pieds et des sensations dans mes pieds ; à chaque fois que je vais le voir, il explore mon pied à l’aide d’une aiguille pour voir si je perds des réflexes ou des sensations. Parfois, la présence d’une barre d’appui ou d’une autre chose de ce genre me rassure. Je ne peux pas précisément dire pourquoi ; je pense qu'à ce stade, c’est plus un réflexe qu'un besoin ou une nécessité. À moins que je me mente à moi-même et que cela commence à devenir une nécessité. Je suis certain d’avoir perdu beaucoup de force dans les mains. Je ne peux plus faire certaines choses, comme enclencher un interrupteur, à moins de fournir de très gros efforts ou d’utiliser 2 doigts, ou de m’aider d’un autre doigt par exemple.
Mon cerveau n’est peut-être plus aussi rapide ni aussi vif. Il est aussi très clair que j’ai des troubles de mémoire. C’est peut être un exemple idiot, mais il m’arrive de courir autour de la maison en pensant à 3 tâches. Je commence, je fais deux choses, et j'oublie ce qu’était la troisième. Alors il faut que je revienne en arrière et que je refasse le processus en quelle que sorte. Dans un travail d’analyse, comme celui qu’est censé faire un avocat, à chaque fois que je lis quelque chose sur une affaire, je passe à côté d’éléments qui ne m’auraient pas échappé, qui me seraient presque automatiquement venus à l’esprit auparavant. Je ne sais pas si c'est l’alcool ou autre chose, ou juste l’âge. C’est difficile à dire parce que c’est très irrégulier.
AU CARREFOUR : QUESTIONS AU DR BRUST
Comment l'alcool affecte-t-il le système nerveux et que sait-on de la prévalence de ces atteintes ? Quelles sont les principales causes et orientations diagnostiques à envisager lors de l’évaluation d’anomalies du système nerveux chez un homme de 74 ans qui est un buveur actif ? Comment détermine-t-on si les symptômes ou les signes sont liés à l'alcool ? En ce qui concerne la cognition, y a-t-il un seuil de « dose inoffensive » d’alcool ? Que recommandez-vous pour M. E. ?
DR BRUST : Depuis plus d’une dizaine d’années, M. E. présente une polyneuropathie sensitivomotrice lentement progressive, de type démyélinisant mais d'étiologie indéterminée. Il y a plusieurs années, son taux sérique limite de cobalamine a été traité par supplémentation en vitamine B12. Au cours des dernières années, il a présenté des troubles de la mémoire récente - oubliant ce qu'il allait dire, hésitant à trouver ses mots, et contraint d’établir des listes. Après sa retraite il y a plus de dix ans, il a progressivement augmenté sa consommation d'alcool jusqu’à un degré qu'il reconnaît comme excessif. Parmi les incertitudes diagnostiques figure la question de savoir si l'alcool contribue à sa polyneuropathie ou à ses troubles cognitifs.
Effets de l'alcool sur le système nerveux
L'alcool endommage le système nerveux d’innombrables manières, et bien que la prévalence de ces complications soit difficile à évaluer, elle est probablement sous-estimée (ENCADRÉ 1). La carence en thiamine, et probablement en d’autres vitamines, conduit ou contribue fortement au syndrome de Wernicke-Korsakoff, à la dégénérescence cérébelleuse, à la polyneuropathie, et à l’amblyopie. La pellagre, qui entraîne un déficit cognitif et des symptômes psychiatriques, résulte d’une déficience en acide nicotinique. Dans le cas de M. E., la polyneuropathie alcoolique/nutritionnelle a un caractère essentiellement axonal, avec des vitesses de conduction nerveuse seulement modestement réduites.3
Encadré 1. Complications neurologiques d’une forte consommation d’alcool- Effets nutritionnels
- Syndrome de Wernicke-Korsakoff1
- Dégénérescence cellulaire2
- Polyneuropathie3
- Amblyopie4
- Pellagre5
- Effets indirects
- Encéphalopathie hépatique6
- Hypoglycémie7
- Acidocétose alcoolique8
- Infection9
- Cancer10
- Traumatisme11
- Accident vasculaire cérébrale12
- Effets directs ou moins bien compris
- Myopathie13
- Maladie de Marchiafava-Bignami14
- Démence alcoolique15
- Syndrome d’alcoolisme foetal16
|
|
L’un des symptômes précoces de sa polyneuropathie était la faiblesse, qui était suffisamment saillante pour soutenir un diagnostic provisoire de neuropathie sensitivomotrice héréditaire de type CMT.17 La polyneuropathie chez les personnes alcooliques, en revanche, produit une perte sensitive précoce, souvent douloureuse, la faiblesse étant habituellement un signe tardif. En outre, les symptômes névropathiques de M. E. sont apparus des années avant qu'il ait commencé à boire excessivement, et son histoire n'indique pas de carence nutritionnelle. Par ailleurs, la toxicité directe de l’alcool contribue probablement à la polyneuropathie de nombreux grands buveurs, et il est théoriquement possible qu’elle ait pu aggraver les symptômes névropathiques de M. E.
À l'examen, M. E. présentait une anomalie de la marche avec élargissement du polygone de sustentation, ce qui peut résulter d’une polyneuropathie, notamment en cas d’altération de la sensibilité proprioceptive. Comme la faiblesse, le déficit proprioceptif tend à être une manifestation tardive de la polyneuropathie dans l'alcoolisme. La démarche et l'ataxie tronculaire chez ces patients sont le plus souvent attribuables à une dégénérescence du vermis cérébelleux (qui, comme la polyneuropathie, résulte généralement d’une carence nutritionnelle et d’une neurotoxicité directe).18
Indirectement, l'alcool peut endommager le système nerveux en causant ou en augmentant le risque d'insuffisance hépatique, d’hypoglycémie, d’acidocétose alcoolique, d'infection, de néoplasme, de traumatisme, et d’accident vasculaire cérébral (ENCADRÉ 1). Chacun de ces troubles peut induire un déficit cognitif, mais aucun élément n’est suggéré dans l’histoire de M. E.
La myopathie alcoolique, la maladie de Marchiafava-Bignami (une leucoencéphalopathie mal comprise affectant particulièrement le corps calleux), la démence alcoolique non-nutritionnelle, et les effets de l'alcool sur le fœtus (ENCADRÉ 1) sont moins bien connus. La neurotoxicité directe semble jouer un rôle majeur dans ces troubles.
Déficit cognitif léger et maladie d'Alzheimer
Les principales orientations diagnostiques chez un grand buveur de 74 ans présentant des pertes de mémoire sont le déficit cognitif léger (MCI) lié à l’âge, la maladie d'Alzheimer et d’autres troubles démentiels, la dépression, et les effets de l'alcool. Comment les cliniciens les distinguent-ils ?
Le déficit cognitif léger se réfère à une altération de la fonction cognitive, typiquement de la mémoire, dans une plus grande mesure qu’attendu pour l'âge mais sans atteindre la démence, définie par le DSM-IV comme un déficit cognitif affectant plus que la mémoire et suffisamment sévère pour interférer avec le fonctionnement professionnel ou social.19 Un certain degré de déclin cognitif peut être partie du vieillissement normal, et la maladie d'Alzheimer débute insidieusement, le plus souvent par des pertes de mémoire subtiles. D’importantes recherches ont donc été consacrées à la définition des limites du MCI (où finit le vieillissement normal et où commence la démence d'Alzheimer), mais aucun consensus concernant les critères spécifiques du MCI (donc de sa prévalence) n’a encore été obtenu.20,21
Avant que la maladie d'Alzheimer produise les symptômes répondant aux critères de démence apparaissent des déficits dans plus d’un domaine cognitif, y compris l’attention, le langage, le calcul, l’orientation spatiale, la fonction exécutive, et la capacité d’abstraction. Certains chercheurs divisent le MCI en sous-groupes basés sur la présence d’une altération de la mémoire (amnésique) sans autre dysfonctionnement cognitif, d’une altération de la mémoire plus autre dysfonctionnement cognitif, ou de tout autre dysfonctionnement cognitif sans altération de la mémoire et impliquant 1 domaine cognitif non-amnésique ou plus.22 Dans 1 grande série, le MCI non-amnésique était aussi fréquent que le MCI amnésique.23 L'atteinte de plus d’un domaine cognitif ne peut donc pas être utilisée pour distinguer le MCI et la maladie d'Alzheimer précoce.
En fonction de différents critères employés pour définir le MCI, des études de cohorte ont rapporté un taux d’évolution vers la démence entre 6 % et 25 % par an, la plupart rapportant des taux situés entre 10 % et 15 %.20,24-28 La progression vers la démence, le plus souvent la maladie d'Alzheimer, est de près de 80 % en l’espace de plusieurs années. Dans l'étude LEILA 75+ (Leipzig Longitudinal Study of the Aged) qui évaluait 980 individus vivant dans la communauté, âgés de 75 ans ou plus, le diagnostic de MCI était basé sur le rapport subjectif de déficit cognitif, sur la capacité préservée d'exercer des activités de la vie quotidienne, et sur l'absence de démence selon les critères du DSM-IV.23 À l’inclusion, 19,2 % des participants avaient un MCI (4,5 % amnésiques dans un seul domaine, 5,5 % amnésiques dans des domaines multiples, 7,1 % non-amnésiques dans un seul domaine, et 2,1 % non-amnésiques avec atteinte multi-domaines). Lors d’un suivi moyen de 4,3 ans, 44 % des individus avec un diagnostic de MCI ont développé une démence comparé à 17 % chez ceux n’ayant pas de déficit cognitif. Les patients avec un MCI amnésique avaient une plus grande probabilité de progresser vers la démence que ceux avec un type non-amnésique. Dans l'étude Cardiovascular Health Study Cognition Study, le MCI amnésique chez les individus avec des troubles associés (psychiatriques, neurologiques, ou systémiques) pouvant causer des déficits cognitifs était moins susceptible de progresser vers la démence que chez ceux sans comorbidité (41 % vs 70 %).29 Des études épidémiologiques utilisant des critères variables ont constaté, chez 4 % à 44 % des individus avec MCI, non seulement une absence de progression vers la démence mais également le retour à une cognition normale.23,29-32
L'examen neuropsychologique permet d’identifier la présence, le type, et le degré d'altération cognitive, et d’évaluer les variations de la cognition au fil du temps ; cependant, aux stades précoces du MCI, il ne permet pas de prédire si les symptômes progresseront vers la démence.20 Certains investigateurs ont décrit l'utilité de la neuroimagerie, des études électrophysiologiques ou des biomarqueurs, mais ces technologies ne sont pas largement disponibles et leur rôle clinique n'est pas encore connu (ENCADRÉ 2).33-38 Actuellement, il est difficile de prédire l’évolution du MCI chez un individu.
| Encadré 2. Études de neuro-imagerie, électrophysiologiques, et des biomarqueurs dans l’altération cognitive (MCI) a Image de résonance magnétique mesurant l’hippocampe et l’atrophie entorhinale : Pendant un suivi de 1 à 9 ans, les patients ayant une MCI et de plus petits volumes de cortex avaient plus de probabilité de progresser vers une maladie d'Alzheimer que les patients ayant une MCI et de plus grands volumes ou que les témoins sains sur le plan cognitif avec des volumes normaux (P<0.001).33
Tomographie d'émission de positron, utilisant FDDNP, une molécule radio-traceur qui se lie à l'amyloïde et à la protéine tau : Une liaison sensiblement plus élevée se produit chez les patients ayant une maladie d'Alzheimer que chez les patients ayant une MCI et chez ceux patients ayant une MCI que chez les témoins sains sur le plan cognitif (P<0.001).34
Mesure encéphalographic magnétique de la densité pariétale gauche des dipôles delta (LPD) : Les patients ayant une MCI avec une forte LPD avaient plus de probabilité de développer une maladie d'Alzheimer dans un délai de 2 ans que ceux ayant une LPD basse (P=0.002).35
Evaluation (ERP) potentielle liée aux événements d'un composant ERP positif négatif durant la mémoire de travail (ERPWM) : Les patients MCI sans déclin cognitif pendant un suivi d'une année avaient à la ligne de base des ERP-WM visibles. Les patients ayant une MCI avec déclin cognitif progressif n’en avaient pas (valeur de P non donnée).36
Mesure des niveaux plasmatiques de l'amyloid β-protéine 42 : Les niveaux chez les femmes ayant une MCI étaient sensiblement plus élevés que chez les femmes en bonne santé sur le plan cognitif (P<0.05)37 ou les hommes ayant une MCI (P<0.002).
Mesure de la protéine tau dans le liquide céphalo-rachidien : Les patients ayant une MCI présentant des niveaux élevés de protéine tau à la ligne de base avaient plus de probabilité d’avoir une détérioration cognitive au cours d’un suivi de deux ans que les patients ayant une MCI présentant des niveaux normaux de tau.38
aLes études n’ont pas été établies pour une utilisation systématique en clinique.
|
|
Autres troubles démentiels chez les personnes âgées
Chez les patients présentant un MCI ou une démence vraie, des diagnostics autres que la maladie d'Alzheimer doivent être envisagés, en particulier ceux qui peuvent être traités. Les causes les plus communes de démence chez les personnes âgées après la maladie d'Alzheimer sont la démence vasculaire, la démence à corps de Lewy/maladie de Parkinson, l’alcoolisme, et l’intoxication par des drogues ou des médicaments. Dans une étude britannique de 1 085 personnes âgées de 65 ans ou plus, la démence était présente chez environ 10 %, sur lesquels deux-tiers ont reçu un diagnostic spécifique. Trente et un pour cent avaient une maladie d'Alzheimer probable, 22 % une démence vasculaire, et 11 % une démence à corps de Lewy.39 Une revue de la littérature a rapporté une prévalence de la démence à corps de Lewy de 0 % à 5 % dans la population générale, et de 0 % à 35 % parmi tous les cas de démence.40
La démence par infarctus multiples se réfère au déclin cognitif résultant d’accidents cérébrovasculaires répétés de gros vaisseaux, impliquant généralement plusieurs régions cérébrales. L'histoire est généralement ponctuée d’épisodes discrets de troubles neurologiques soudains. M. E. ne présente pas ces caractéristiques. En dehors de sa consommation excessive d'alcool, l'hypertension est son seul facteur de risque connu d’AVC. Ses symptômes pourraient être liés à une microangiopathie diffuse touchant de petites artères et artérioles cérébrales perforantes. Les changements ischémiques de ce type sont reflétés dans de multiples zones souvent confluentes, qui ont un signal de haute intensité anormal à l’IRM avec séquence pondérée en T2 ou FLAIR (fluid-attenuated inversion recovery) (appelées leucoaraïoses), impliquant la substance blanche périventriculaire et la couronne rayonnante.41 Dans cette forme de démence vasculaire, parfois appelée maladie de Binswanger, le déclin cognitif peut être insidieux dans le début et lentement progressif. Des signes moteurs et des troubles de la marche accompagnent souvent la démence.42
Le rapport entre l’alcoolisme et les maladies cérébrovasculaires est complexe. Comme dans les coronaropathies, la consommation d’alcool faible à modérée semble réduire le risque d’accident ischémique comparé à l'abstinence, tandis que la forte consommation en majore le risque. Le rapport produit ainsi une « courbe en J ».12 Il existe une relation similaire en J pour la consommation d'alcool et les anomalies de la substance blanche cérébrale à l’IRM.43
La démence peut être une caractéristique de la maladie de Parkinson, survenant généralement tardivement au cours de la maladie. Avec la maladie à corps de Lewy, le déclin cognitif, souvent accompagné de signes psychiatriques comprenant des hallucinations, peut précéder les symptômes de parkinsonisme.44 La question de savoir si la maladie de Parkinson et la démence à corps de Lewy représentent des entités distinctes ou un continuum est controversée ; cependant 10 % à 30 % des patients âgés avec démence présentent des corps de Lewy corticaux à l’autopsie.44,45 Le déclin cognitif de M. E. n'inclut pas de caractéristiques évocatrices de la maladie à corps de Lewy, et son examen, y compris sa marche avec élargissement du polygone de sustentation, ne suggère pas de parkinsonisme.
Les effets médicamenteux doivent être envisagés chez un patient âgé présentant des symptômes cognitifs subtils. M. E. prend du vérapamil et de la ranitidine qui peuvent tous deux avoir des effets indésirables cognitifs,46,47 et de l’hydrochlorothiazide qui peut produire indirectement des symptômes mentaux liés à une hyponatrémie ou à une hypercalcémie. Aucun de ces médicaments ne constituerait une explication probable de ses troubles de mémoire ; cependant une corrélation temporelle entre ses symptômes et l’initiation d'un médicament particulier ou une modification posologique doit être recherchée.
Les démences curables chez les individus âgés incluent un certain nombre de troubles métaboliques/systémiques et d’atteintes primaires du SNC (ENCADRÉ 3). Dans le cas de M. E., une carence en cobalamine pourrait être impliquée ; elle survient chez environ 10 % de la population américaine de plus de 70 ans48 et peut causer des symptômes neurologiques en l'absence d'anémie, y compris un changement cognitif et une polyneuropathie démyélinisante.49 Cependant, cela serait insolite en l’absence de signe de myélopathie ; en outre, le traitement par vitamine B12 n’a pas amélioré ses symptômes neurologiques.50
Encadré 3. Causes traitables de démence- Troubles métaboliques/systémiques
- Déficit en cobalamine, thiamine, ou niacine
- Hypothyroïdie ou hyperthyroïdie
- Insuffisance surrénalienne ou Excès surrénalien
- Hypocalcémie ou hypercalcémie
- Insuffisance rénale, hépatique ou pulmonaire
- Intoxication
- Hématome chronique sous-dural
- Hydrocéphalie à pression normale
- Infection chronique du système nerveux central
- Tumeur cérébrale
- Crises non convulsives
- Dépression
|
|
La combinaison de la démarche instable et de l’altération de l’état mental de M. E. pourrait suggérer une hydrocéphalie à pression normale. Cependant, dans cette affection, les troubles de la marche sont généralement précoces et prédominants, et le déclin cognitif se compose plus souvent d’une apathie mentale (aboulie) que d’une légère perte mnésique.51
La pseudodémence dépressive, autrefois considérée comme un artefact d’une vitesse de traitement diminuée, se révèle être un véritable déficit cognitif. Les personnes âgées non démentes déprimées ont des volumes hippocampiques réduits, et dans des modèles animaux de dépression, les neurones hippocampiques cessent de se renouveler.52-54 La forte consommation d'alcool et la dépression sont généralement associées, bien que l'alcoolisme soit plus souvent la cause que le résultat de la dépression.55 Monsieur E. ne se décrit pas comme déprimé, mais il a augmenté sa consommation d'alcool après sa retraite, quand il « broyait du noir », et dans les moments où il était isolé de sa famille. L’interrogatoire chez ces patients doit inclure une recherche de la dépression, incluant des questions sur la déprime, l'insomnie, le manque d'énergie, et la perte d'appétit.56
Syndrome de Wernicke-Korsakoff et démence alcoolique
Bien que pathologiquement semblables et causés par une carence en thiamine, les syndromes de Wernicke et Korsakoff sont cliniquement distincts. Évoluant pendant des jours à des semaines, le syndrome de Wernicke se compose de la triade incluant altération de l’état mental, troubles oculomoteurs, et démarche ataxique. Les anomalies mentales incluent la léthargie, la distraction, l'aboulie, et l’altération de la mémoire, qui progressent vers le coma en l’absence de traitement. La restriction des mouvements oculaires évolue vers une ophthalmoplégie complète. L'ataxie tronculaire peut empêcher la station debout ou la marche. Non traitée, cette affection est mortelle ; pathologiquement, des lésions histologiquement distinctes sont observées dans le thalamus médial et l'hypothalamus, la substance grise péri-épendymaire du mésencéphale, et le pont. Après supplémentation thiaminique et multivitaminique, les symptômes commencent à s'améliorer dans les heures ou les jours qui suivent, mais le syndrome de Korsakoff persiste chez plus de la moitié des patients traités. Dans cette phase de la maladie, la vigilance, la capacité d’attention, le comportement, et la mémoire de travail (conservation > 30 secondes) sont relativement préservés, mais il y a une amnésie durable et souvent sévère, tant antérograde, avec incapacité de retenir des informations nouvelles, que rétrograde, avec perte de la mémoire d'événements anciens, datant de mois ou d’années. L’absence de conscience du trouble est souvent observée ; la fabulation peut être présente ou absente. L'examen psychologique révèle un déficit cognitif inexpliqué par la perte de mémoire pure, mais l'amnésie prédomine.57,58
La prévalence du syndrome de Wernicke-Korsakoff est indéterminée, car le syndrome de Wernicke est souvent non reconnu cliniquement, en particulier quand les troubles oculomoteurs n'accompagnent pas le changement mental. Dans une étude australienne, sur 131 cas diagnostiqués à l'autopsie comme ayant l'encéphalopathie de Wernicke, seulement 26 (20 %) avaient été diagnostiqués cliniquement.59 Dans une étude de 22 neuropathologistes d'Australie, d'Autriche, de Belgique, de Tchéquie, de France, d'Allemagne, de Norvège, du Royaume-Uni, et des États-Unis, la prévalence du syndrome de Wernicke-Korsakoff à l'autopsie variait de 0 % à 2,8 %.60 Il est intéressant de noter que la prévalence de ce syndrome n’était pas corrélée à la quantité moyenne d'alcool consommée dans chaque pays. La France, avec une consommation d’alcool moyenne annuelle par habitant de 12,8 l, avait une prévalence de 0,4 % à 1,3 %. L'Australie, avec une consommation moyenne de 8,4 l, avait une prévalence de 2,1 % à 2,8 %.
La fréquence à laquelle survient le syndrome de Korsakoff sans précéder le syndrome de Wernicke et la fréquence à laquelle les troubles mentaux sont cliniquement limités à la mémoire sont des questions difficiles à résoudre sachant que l'alcool éthylique a une neurotoxicité directe sur les systèmes nerveux central et périphérique, et que cette neurotoxicité peut induire un changement cognitif progressant vers la démence en l'absence de carence nutritionnelle, d’encéphalopathie hépatique, de traumatisme cérébral, ou d'autres causes indirectes. Dans des études animales utilisant des témoins avec alimentation appariée, l’administration chronique d'éthanol a induit une perte neuronale dans plusieurs régions cérébrales incluant l’hippocampe, le prosencéphale basal, le cortex cérébral, et le cervelet.61-64 L’alcoolisme périodique est particulièrement susceptible de produire des lésions cérébrales.65 Les atteintes plus importantes conférées par des périodes répétées de consommation et de sevrage, comparé à un alcoolisme continu, suggèrent un mécanisme physiopathologique potentiel. Plus précisément, l'alcool éthylique inhibe la neurotransmission excitatrice par le glutamate, avec une exagération de la transmission glutamatergique au cours du sevrage.66 Les périodes répétées de sevrage pourraient ainsi exposer les neurones à une excitotoxicité et à un stress oxydatif.67
Chez l'homme, l’alcoolisme excessif est associé à une dilatation des espaces ventriculaires, à un élargissement des sillons corticaux, et à une anomalie des signaux de la substance blanche cérébrale à l’imagerie par résonance magnétique.68,69 Pathologiquement, une perte neuronale se produit dans le cortex cérébral frontal, le thalamus, l'hypothalamus, le cervelet, et, moins régulièrement, dans l’hippocampe.70 Les lésions du lobe frontal sont corrélées à des troubles cognitifs non-amnésiques tels que les difficultés de planification, d’organisation, de résolution des problèmes, et d’abstraction, ainsi qu’à la persistance des réponses et à la désinhibition.71
Il est à noter que l'abstinence soutenue chez ces patients peut inverser l’élargissement des ventricules et des sillons.72,73 Une étude utilisant la spectroscopie par résonance magnétique a démontré que l'amélioration morphologique était corrélée à une augmentation du taux de choline (marqueur de myélinisation) dans la substance blanche et de N-acétyl-aspartate (marqueur neuronal) dans le cortex frontal. Des taux supérieurs de N-acétyl-aspartate ont été associés à une amélioration de la cognition.74 Cependant, le retour à la normale de la cognition n’a pas été établi.
La maladie d'Alzheimer et la maladie ischémique coexistent souvent chez un même patient ; la part contributive de chacune au déficit cognitif ne peut donc être établie avec certitude. La tâche devient encore plus complexe si l’on ajoute à l'équation les effets indésirables d'une forte consommation d'éthanol sur la cognition et la maladie cérébrovasculaire.
Effet neuroprotecteur de l’alcool
Si l'alcool est neurotoxique, il est important de définir un seuil de dose inoffensive. Un certain nombre d'études épidémiologiques n'ont pas pu identifier d’association positive entre la consommation modérée d'alcool (généralement jusqu'à 1 ou 2 verres par jour) et le risque de démence, et ont en outre trouvé que ces quantités avaient un effet protecteur.75-83 Certaines études ont démontré un effet protecteur spécifique du vin.79,80,82 Une consommation plus élevée augmentait le risque de démence. Les effets de l'alcool sur la cognition sont donc similaires à ses effets sur l’ischémie vasculaire, produisant une courbe en forme de J, les doses faibles à modérées réduisant le risque et les fortes doses le majorant. Cependant, les effets cérébrovasculaires n'expliquent pas entièrement les effets protecteurs de l'alcool sur la cognition. Les propriétés antioxydantes des boissons alcoolisées ont été suggérées comme mécanisme potentiel.84
Une récente revue comparant les effets protecteurs allégués de la consommation d’alcool faible à modérée sur la coronaropathie et la mortalité toutes causes confondues pourrait être en rapport avec le bénéfice apparent de la consommation d’alcool faible à modérée sur la cognition et l’accident ischémique. En examinant 54 études épidémiologiques qui rapportaient un tel bénéfice, les analyseurs ont constaté que beaucoup avaient comparé des personnes avec une consommation d’alcool faible à modérée avec un groupe témoin composé d’anciens buveurs et de personnes n’ayant jamais bu. Les auteurs ont supposé que les anciens buveurs incluaient probablement des individus ayant cessé de boire pour des raisons de santé, et, en effet, quand les études utilisant ce groupe de comparaison ont été ôtées de l'analyse, le bénéfice de la consommation faible à modérée d'alcool n'était plus visible.85 Il reste à voir si les effets rapportés de l'alcool sur la cognition et les maladies cérébrovasculaires persisteront après une analyse méthodologique similaire.
Mesures de prise en charge
Compte tenu des données qui précèdent, quelles sont les solutions de prise en charge pour Monsieur E. ? Une première étape consisterait à documenter le degré et la nature de son déficit cognitif. Le MMSE (Mini-Mental State Examination) est un test de la fonction cognitive en 30 points, qui évalue l'orientation, l'attention, la mémoire immédiate et de travail, le langage, le calcul, et les praxies constructives.86 Cependant, il n’est pas sensible chez les patients présentant un MCI ou la maladie d'Alzheimer précoce, chez lesquels les inquiétudes subjectives sur l’altération de la mémoire peuvent ne pas être reflétées dans les tests d’orientation ou de rappel des mots ; en outre, il n'est pas conçu pour dépister la dépression, le dysfonctionnement exécutif, ou les troubles du comportement. Chez les individus ayant un niveau d’instruction supérieur, les seuils des scores définissant les anomalies sont généralement ajustés vers le haut.87 Le MMSE a une sensibilité et une spécificité acceptables dans le dépistage de la démence, mais des examens neuropsychologiques plus formels et plus étendus peuvent être nécessaires pour documenter un dysfonctionnement cognitif subtil, en particulier quand le trouble mnésique n'est pas prédominant.88
L’étape suivante consisterait à exclure d'autres causes de trouble cognitif potentiellement curables (voire réversibles), une tâche généralement effectuée par l’imagerie cérébrale et quelques tests biologiques (ENCADRÉ 2). Dans une revue de 9 séries cliniques incluant 800 individus déments âgés de 65 ans ou plus, 82 avaient une démence « curable » (neurosyphilis, infection fongique, tumeur cérébrale, alcoolisme, hématome sous-dural, hydrocéphalie, épilepsie) et 53 avaient une démence « réversible » (toxicité médicamenteuse, trouble « métabolique », hépatique, hyponatrémie, hyper ou hypocalcémie, carence en vitamine B12, maladie thyroïdienne, hypoglycémie).89 Dans une autre série de 200 individus atteints de démence, 9,5 % avaient une « démence induite par une substance », 4 % une « démence liée à l’alcool », 3 % un hypothyroïdisme, 1 % un hyperparathyroïdisme, 1 % une hyponatrémie, et 0,5 % une hypoglycémie.90
L’hypothèse de travail pour Monsieur E. sera probablement le MCI ou la maladie d'Alzheimer précoce, une polyneuropathie indépendante, et une incertitude quant au rôle de l'alcool. Compte tenu de la possibilité d’une aggravation des symptômes induite par l'alcool - et il n’est pas déraisonnable de supposer que chez les individus avec MCI ou polyneuropathie, une petite quantité d'alcool pourrait avoir davantage de portée, c’est-à-dire avoir des effets plus délétères – l’abstinence doit être recommandée. Monsieur. E pourrait devoir participer à un programme comme les Alcooliques Anonymes ou être traité par l’un des 3 médicaments approuvés par la FDA américaine (US Food and Drug Administration) pour le traitement de l'alcoolisme chronique (disulfirame, naltrexone, ou acamprosate). En cas de persistance de l’alcoolisme, une supplémentation thiaminique et multivitaminique serait indiquée.
Quant au traitement du MCI, une étude en double aveugle, contrôlée contre placebo, a démontré que le donépézil, inhibiteur de la cholinestérase, réduisait la probabilité de progression vers la maladie d'Alzheimer pendant les 12 premiers mois de l'étude. Le bénéfice, cependant, semble porter plutôt sur les symptômes que sur un ralentissement de la progression de la maladie ; après 3 ans, les patients recevant le donépézil ou le placebo avaient la même probabilité de développer la démence d'Alzheimer. Il est intéressant de noter que pour les porteurs de l'allèle  4 de l'apolipoprotéine E, un bénéfice était observé tout au long du suivi de 3 ans.91 Des études bien conçues ont également démontré que les activités de loisirs cognitives (lecture, écriture, mots croisés, jeux de société ou de cartes, discussions de groupe, ou pratique de la musique, excluant les activités physiques) réduisent le risque de développer un MCI ou une démence chez les personnes âgées.92-95 (Dans l’une de ces études, regarder la télévision a été associé à un risque majoré de déficit cognitif.96,97). Certaines données démontrent également que des séances d’entraînement cognitif spécifiques peuvent réduire le risque de déclin cognitif. En dépit des inquiétudes subjectives de M. E concernant ses troubles de mémoire, il semble avoir des capacités mentales plus que suffisantes pour pratiquer ce type d’activité. 4 de l'apolipoprotéine E, un bénéfice était observé tout au long du suivi de 3 ans.91 Des études bien conçues ont également démontré que les activités de loisirs cognitives (lecture, écriture, mots croisés, jeux de société ou de cartes, discussions de groupe, ou pratique de la musique, excluant les activités physiques) réduisent le risque de développer un MCI ou une démence chez les personnes âgées.92-95 (Dans l’une de ces études, regarder la télévision a été associé à un risque majoré de déficit cognitif.96,97). Certaines données démontrent également que des séances d’entraînement cognitif spécifiques peuvent réduire le risque de déclin cognitif. En dépit des inquiétudes subjectives de M. E concernant ses troubles de mémoire, il semble avoir des capacités mentales plus que suffisantes pour pratiquer ce type d’activité.
QUESTIONS ET COMMENTAIRE
QUESTION : Pourquoi avez-vous exclu la dégénérescence cérébelleuse alcoolique, ou du moins l’avez-vous mise au bas de la liste ? Monsieur E. se plaignait de problèmes manuels ; il ne pouvait plus utiliser ses mains. Eugene O'Neill, le dramaturge américain, a été l'un des 10 premiers cas de dégénérescence cérébelleuse alcoolique décrits par Raymond Adams à l'hôpital de Boston. O'Neill se plaignait de ne plus pouvoir tenir un stylo, il ne pouvait pas écrire, il ne pouvait pas terminer ses pièces, et c'était très pénible pour lui. Vous n'avez pourtant pas placé ce syndrome en tête de liste pour ce patient ?
DR BRUST : Je n'ai pas exclu la dégénérescence cérébelleuse alcoolique ; je pense simplement que le principal facteur contribuant aux troubles de la marche de M. E. est sa polyneuropathie. La polyneuropathie peut affecter la sensibilité proprioceptive de ses pieds, entraînant ce que l’on appelle une ataxie sensitive ; son instabilité pourrait également être en grande partie due à la faiblesse. Comme pour sa faiblesse de la main, une paralysie ulnaire surajoutée a été documentée par électromyogramme et est évidente à l'examen, avec la difformité en griffe des quatrième et cinquième doigts.
Par ailleurs, un rapport d'autopsie effectué il y a plusieurs années par Price et Richardson98 a révélé qu'Eugene O'Neill était probablement atteint d’une maladie dégénérative cérébelleuse non-alcoolique/non-nutritionnelle héréditaire ou sporadique. Pendant les 25 dernières années de sa vie, O'Neill ne buvait pas excessivement d'alcool, et il a été abstinent pendant ses 8 dernières années ; pourtant l'ataxie cérébelleuse affectant sa démarche, ses membres, et ses articulations, a progressé implacablement autour de l'âge de 50 ans jusqu'à sa mort, à 65 ans. Ses bras et ses mains étaient si gravement atteints qu'il ne pouvait plus écrire. La dégénérescence cérébelleuse alcoolique tend à causer une ataxie tronculaire tout en épargnant les membres, et en particulier les bras ; les patients concernés sont peu susceptibles de présenter des tremblements des membres supérieurs (sauf en cas de phénomène de sevrage), une dysmétrie, ou une dysdiadochokinésie.99
La dégénérescence cérébelleuse alcoolique peut survenir sans autres signes du syndrome de Wernicke-Korsakoff,99 et je ne peux pas l'exclure en tant que facteur contributif potentiel dans les troubles de la marche de M. E. (qui jusqu’à récemment étaient suffisamment bénins pour lui permettre de jouer au tennis). Cependant, je ne pense pas que nous ayons besoin de la dégénérescence cérébelleuse alcoolique ou de la polyneuropathie alcoolique pour expliquer ses symptômes.
QUESTION : Il est très facile pour nous de dire qu'il devrait s'abstenir de boire, mais il est beaucoup plus difficile de l’imposer en tant que fournisseur de soins primaires. Si l’on en croit le patient, il a consommé peu d’alcool pendant des dizaines d’années, puis, dans un intervalle relativement court, peut-être de dix ans, il est passé à une forte consommation. Existe-t-il un niveau auquel l'alcool est sans danger pour les patients âgés ? Doit-il s'abstenir complètement ou plutôt réduire sa consommation en dessous d'un seuil considéré comme excessif ?
DR BRUST : Ceci entre dans la définition épineuse de l’alcoolisme. C'est un vaste spectre, qui englobe les personnes physiquement dépendantes présentant des tremblements et le delirium tremens quand elles s'arrêtent, et celles qui ne boivent pas quotidiennement mais ont de gros problèmes quand elles boivent. Je pense cependant que pour les personnes qui boivent tous les jours (ce qui est son cas), il est très risqué d’essayer d’être un buveur mondain normal.
Il y a un phénomène appelé la sensibilisation, qui est actuellement un thème d'actualité brûlant dans la recherche sur l’abus de substances et chez les animaux, le plus clairement observé avec les psychostimulants. Selon le schéma posologique, on atteint un point auquel, après une période d'abstinence, l'exposition à la drogue déclenche une réponse excessive (« tolérance inversée ») et un besoin impérieux.100 Ce phénomène pourrait expliquer pourquoi l'alcoolique abstinent pendant des semaines ou des mois ne peut pas s’arrêter s’il reprend 1 verre. Les Alcooliques Anonymes ont toujours reconnu ce danger et sont catégoriques sur le fait que les buveurs à problème doivent observer une abstinence complète. Un verre, ou même le fait d’être dans un environnement associé à la boisson, peut déclencher une sensibilisation et un besoin impérieux – de même que les chiens de Pavlov salivant au son de la clochette.101 Je recommande donc l'abstinence, quoiqu'elle soit rarement obtenue. Le traitement de l'alcoolisme est l'une des tâches les plus difficiles que nous ayons à traiter.
QUESTION : Y a-t-il un niveau d'alcool inoffensif pour le cerveau vieillissant ?
DR BRUST : Dans un cerveau vieillissant avec des troubles cognitifs ? Je ne sais pas. Il est possible que de faibles doses d'alcool soient protectrices dans un cerveau normal et puissent même prévenir le MCI ou la maladie d'Alzheimer. Mais si vous présentez déjà des atteintes, de petites quantités d'alcool peuvent-elles empirer les choses ? Je ne connais aucune donnée dans un sens ou dans l’autre en la matière.
QUESTION : Nous disons aux personnes atteintes de maladie cardiovasculaire que si elles arrêtent de fumer, elles pourront connaître certains bénéfices dès 2 ans ou à peu près, ce qui ramènera leurs risques à ceux d'un non-fumeur en matière d’événement cardiovasculaire aigu. Pouvons-nous dire à ce patient qu'il pourrait connaître des bénéfices ou une amélioration similaires de son déficit cognitif, s’il devait arrêter de boire ?
DR BRUST : Si son déficit cognitif est entièrement attribuable à l'alcool, il pourrait présenter une amélioration. L'étude que j'ai citée, qui démontre une amélioration de la cognition associée à l’augmentation des taux de N-acétyl-aspartate, est très prometteuse.74 Si, comme c’est certainement très probable, ce patient présente un léger déficit cognitif indépendant de l'alcool (ou aggravé par l'alcool), il pourrait obtenir une légère amélioration ; il présente cependant un risque de 80 % de voir son MCI évoluer vers la démence au cours des prochaines années.
QUESTION : Concernant les patients ayant connu une inversion de leur MCI, ont-ils pratiqué des exercices cognitifs ou physiques, ou y a-t-il là un autre mécanisme qui entre en jeu ?
DR BRUST : Je ne sais pas. Les études comparant les patients ayant évolué vers la démence à ceux exempts d’évolution ont donné les nombres des groupes mais n'ont pas décrit de profils comportementaux individuels. Eu égard aux bénéfices démontrés des loisirs cognitifs sur la cognition chez les personnes âgées, il est plausible que celles qui ont présenté une amélioration avaient conservé une occupation en faisant des mots croisés ou en jouant de la clarinette.
QUESTION : Je suis un peu dubitatif quant à son histoire de faible consommation quand il était cadre de grandes entreprises. Mon expérience au fil des ans suggère qu’en prenant de l’âge, les pratiquants du « déjeuner à 2 martinis » n’ont souvent plus l’intelligence aussi affûtée que ce que l’on attendrait d’une personne ayant occupé une haute fonction. Est-ce que cela a été étudié dans le temps ?
DR BRUST : Un degré d'instruction plus élevé réduit le risque de développer la maladie d’Alzheimer.102 En outre, une étude de religieuses âgées a évalué « la capacité linguistique » selon la « densité des idées et la complexité grammaticale » d’essais autobiographiques qu'elles avaient écrits plus de 50 ans auparavant. Celles qui avaient obtenu le score le plus bas sur la capacité linguistique à 20 ans étaient les plus susceptibles d’avoir de faibles scores au test cognitif à un âge avancé et, sur celles décédées, d’avoir une maladie d’Alzheimer confirmée neuropathologiquement.103 Les plaques séniles étaient-elles déjà en formation quand elles avaient 20 ans, ou les religieuses âgées non démentes avaient-elles une plus grande capacité de réserve cognitive lors de la formation des plaques ? Je ne connais aucune étude ayant sélectionné des cadres de haut niveau.
Informations sur les auteurs
Correspondance: John C. M. Brust, MD, Department of Neurology, Columbia University College of Physicians and Surgeons, Harlem Hospital Center, 506 Lenox Ave, New York, NY 10037 (jcb2{at}columbia.edu).
Liens financiers : Aucun déclaré.
Financement/Soutien : cet article de Rencontres Cliniques a été rendu possible en partie grâce à une bourse d’un donneur anonyme.
Rôle du sponsor : Le sponsor n'a pas participé au recueil, l'analyse, et l'interprétation des données ou à la préparation, à la revue, ou à l'approbation du manuscrit.
Autres contributions : Nous remercions le patient pour avoir partagé son histoire et pour nous avoir permis de la publier.
Affiliation de l’auteur : Le Dr Brust est professeur de Beurologie clinique à Columbia University College of Physicians and Surgeons, New York, New York.
Rencontres Cliniques du Beth Israel Deaconess Medical Center est produit et publié par Risa B. Burns, MD, Eileen E. Reynolds, MD, and Amy N. Ship, MD. Tom Delbanco, MD, est le rédacteur en chef de cette série.
Cette conférence a eu lieu au cours des Medicine Grand Rounds du Beth Israel Deaconess Medical Center, Boston, Massachusetts, le 1er mars 2007.
Voir aussi page du Patient.
FMC disponible en ligne à www.jamaarchivescme.com et questions p 1080.
Rédactrice en chef de la section Rencontres cliniques: Margaret A. Winker, MD, Deputy Editor.
BIBLIOGRAPHIE
| |
1. Wood B, Currie J. Presentation of acute Wernicke’s encephalopathy and treatment with thiamine. Metab Brain Dis. 1995;10(1):57-72.
PUBMED
2. Phillips SC, Harper CG, Kril J. A quantitative histological study of the cerebellar vermis in alcoholic patients. Brain. 1987;110(pt 2):301-314.
FREE FULL TEXT
3. Koike H, Iijima M, Sugiura M, et al. Alcoholic neuropathy is clinicopathologically distinct from thiamine-deficiency neuropathy. Ann Neurol. 2003;54(1):19-29.
PUBMED
4. Sivyer G.. Evidence of limited repair of brain damage in a patient with alcoholtobacco amblyopia. Med J Aust. 1989;151(9):541.
PUBMED
5. Serdaru M, Hausser-Hauw C, LaPlane D, et al. The clinical spectrum of alcoholic pellagra encephalopathy. Brain. 1988;111(pt 4):829-842.
FREE FULL TEXT
6. Diehl AM. Alcoholic liver disease. Med Clin North Am. 1989;73(4):815-830.
PUBMED
7. Malouf R, Brust JCM. Hypoglycemia: causes, neurological manifestations, and outcome. Ann Neurol. 1985;17(5):421-430.
PUBMED
8. Lien D, Mader TJ. Survival from profound alcohol-related lactic acidosis. J Emerg Med. 1999;17(5):841-846.
PUBMED
9. Cook RT. Alcohol abuse, alcoholism and damage to the immune system—a review. Alcohol Clin Exp Res. 1998;22(9):1927-1942.
PUBMED
10. Seitz HK, Poschl G, Simanowski VA. Alcohol and cancer. Recent Dev Alcohol. 1998;14:67-95.
PUBMED
11. Rivara FP, Koepsell TD, Jurkovich GL, et al. The effects of alcohol abuse on readmission for trauma. JAMA. 1993;270(16):1962-1964.
FREE FULL TEXT
12. Reynolds K, Lewis B, Nolan JDL, et al. Alcohol consumption and risk of stroke: a meta-analysis. JAMA. 2003;289(5):579-588.
FREE FULL TEXT
13. Ferna ndez-Solà J, Junyent JM, Urbana-Marquez A. Alcoholic myopathies. Curr Opin Neurol. 1996;9(5):400-405.
PUBMED
14. Kohler CG, Ances BM, Coleman AR, et al. Marchiafava-Bignami disease: literature review and case report. Neuropsychiatry Neuropsychol Behav Neurol. 2000;13(1):67-76.
PUBMED
15. Harper C, Kril J, Daly J. Are we drinking our neurons away? Br Med J (Clin Res Ed). 1987;294(6571):534-536.
PUBMED
16. Roebuck TM, Mattson SN, Riley EP. A review of the neuroanatomical findings in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol Clin Exp Res. 1998;22(2):339-344.
PUBMED
17. Young P, Suter U. The causes of Charcot-Marie-Tooth Disease. Cell Mol Life Sci. 2003;60(12):2547-2560.
PUBMED
18. Karhunen PJ, Erkinjuntti T, Laippala P. Moderate alcohol consumption and loss of Purkinje cells. BMJ. 1994;308(6945):1663-1667.
FREE FULL TEXT
19. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association; 1994:133-155.20. Petersen RC, Stevens JC, Ganguli M, et al. Practice parameter: early detection of dementia: mild cognitive impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56(9):1133-1142.
FREE FULL TEXT
21. Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol. 2001;58(12):1985-1992.
FREE FULL TEXT
22. Lopez OL, Becker JT, Jagust WJ, et al. Neuropsychological characteristics of mild cognitive impairment subgroups. J Neurol Neurosurg Psychiatry. 2006;77(2):159-165.
FREE FULL TEXT
23. Busse A, Hensel A. Gu hne U, et al.Mild cognitive impairment: long-term course of four clinical subtypes. Neurology. 2006;67(12):2176-2185.
FREE FULL TEXT
24. Devanand DP, Folz M, Gorlyn M, et al. Questionable dementia: clinical course and predictors of outcome. J Am Geriatr Soc. 1997;45(3):321-328.
PUBMED
25. Daly E, Zaitchik D, Copeland M, et al. Predicting progression to Alzheimer’s disease using standardized clinical information. Arch Neurol. 2000;57(5):675-680.
FREE FULL TEXT
26. Bowen J, Teri L, Kukull W, et al. Progression to dementia in patients with isolated memory loss. Lancet. 1997;349(9054):763-765.
PUBMED
27. Flicker C, Ferris SH, Reisberg B. Mild cognitive impairment in the elderly: predictors of dementia. Neurology. 1991;41(7):1006-1009.
FREE FULL TEXT
28. Tuokko H, Frerichs R, Graham J, et al. Five-year follow-up of cognitive impairment with no dementia. Arch Neurol. 2003;60(4):577-582.
FREE FULL TEXT
29. Lopez OL, Kuller LH, Becker JT, et al. Incidence of dementia in mild cognitive impairment in the Cardiovascular Health Study Cognition Study. Arch Neurol. 2007;64(3):416-420.
FREE FULL TEXT
30. Larrieu S, Letenneur L, Orgogozo JM, et al. Incidence and outcome of mild cognitive impairment in a population-based prospective cohort. Neurology. 2002;59(10):1594-1599.
FREE FULL TEXT
31. Solfrizzi V, Panza F, Colacicco AM, et al. Vascular risk factors, incidence of MCI, and rates of progression to dementia. Neurology. 2004;63(10):1882-1891.
FREE FULL TEXT
32. Ganguli M, Dodge HH, Shen C, Dekosky ST. Mild cognitive impairment, amnestic type: an epidemiologic study. Neurology. 2004;63(1):115-121.
FREE FULL TEXT
33. Devanand DP, Pradhaban G, Liu X, et al. Hippocampal and entorhinal atrophy in mild cognitive impairment: prediction of Alzheimer disease. Neurology. 2007;68(11):828-836.
FREE FULL TEXT
34. Small GW, Kepe V, Ercoli LM, et al. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355(25):2652-2663.
PUBMED
35. Ferna’ndez A, Turrero A, Zuluaga P, et al. Magnetoencephalographic parietal delta dipole density in mild cognitive impairment: preliminary results of a method to estimate the risk of developing Alzheimer disease. Arch Neurol. 2006;63(3):427-430.
FREE FULL TEXT
36. Missonnier P, Gold G, Fazio-Costa L, et al. Early event-related potential changes during working memory activation predict rapid decline in mild cognitive impairment. J Gerontol A Biol Sci Med Sci. 2005;60(5):660-666.
FREE FULL TEXT
37. Assini A, Cammarata S, Vitali A, et al. Plasma levels of amyloid beta-protein 42 are increased in women with mild cognitive impairment. Neurology. 2004;63(5):828-831.
FREE FULL TEXT
38. Maruyama M, Matsui T, Tanji A, et al. Cerebrospinal fluid tau protein and periventricular white matter lesions in patients with mild cognitive impairment: implications for 2 major pathways. Arch Neurol. 2004;61(5):716-720.
FREE FULL TEXT
39. Stevens T, Livingston G, Kitchen G, et al. Islington study of dementia subtypes in the community. Br J Psychiatry. 2002;180:270-274.
FREE FULL TEXT
40. Zaccai J, McCracken C, Brayne C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing. 2005;34(6):561-566.
FREE FULL TEXT
41. Leifer D, Buananno FS, Richardson EP. Clinicopathologic correlations of cranial magnetic resonance imaging periventricular white matter. Neurology. 1990;40(6):911-918.
FREE FULL TEXT
42. Mast H, Mohr JP. Binswanger’s disease and vascular dementia. In: Mohr JP, Choi DW, Grotta JC, Weir B, Wolf PA, eds. Stroke: Pathophysiology, Diagnosis, and Management. 4th ed. Philadelphia, PA: Churchill Livingstone; 2004:679-686.43. Mukamal KJ, Longstreth WT, Mittleman MA, et al. Alcohol consumption and subclinical findings on magnetic resonance imaging of the brain in older adults: the Cardiovascular Health Study. Stroke. 2001;32(9):1939-1946.
FREE FULL TEXT
44. Noe E, Marder K, Bell KL, et al. Comparison of dementia with Lewy bodies to Alzheimer’s disease and Parkinson’s disease with dementia. Mov Disord. 2004;19(1):60-67.
PUBMED
45. Bogaerts V, Engelborghs S, Kumar-Singh S, et al. A novel locus for dementia with Lewy bodies: a clinically and genetically heterogeneous disorder. Brain. 2007;130(pt 9):2277-2291.
FREE FULL TEXT
46. Jacobsen FM, Sack DA, James SP. Delirium induced by verapamil. AmJ Psychiatry. 1987;144(2):248.
PUBMED
47. Wormsley KG. Safety profile of ranitidine: a review. Drugs. 1993;46(6):976-985.
PUBMED
48. Stabler SP, Lindenbaum J, Allen RH. Vitamin B12 deficiency in the elderly: current dilemmas. Am J Clin Nutr. 1997;66(4):741-749.
FREE FULL TEXT
49. Healton EB, Savage DG, Brust JCM, et al. Neurologic aspects of cobalamin deficiency. Medicine (Baltimore). 1991;70(4):229-245.
PUBMED
50. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
PUBMED
51. Vanneste JAL. Diagnosis and management of normal-pressure hydrocephalus. J Neurol. 2000;247(1):5-14.
PUBMED
52. Nebes RD, Butters MA, Mulsant BH, et al. Decreased working memory and processing speed mediate cognitive impairment in geriatric depression. Psychol Med. 2000;30(3):679-691.
PUBMED
53. Bell-McGinty S, Butters MA, Meltzer CC, et al. Brain morphometric abnormalities in geriatric depression: long-term neurobiological effects of illness duration. Am J Psychiatry. 2002;159(8):1424-1427.
FREE FULL TEXT
54. Steffens DC, Otley E, Alexopoulis GS, et al. Perspectives on depression, mild cognitive impairment, and cognitive decline. Arch Gen Psychiatry. 2006;63(2):130-138.
FREE FULL TEXT
55. Harrington R, Fudge H, Rutter M, et al. Adult outcomes of childhood and adolescent depression. Arch Gen Psychiatry. 1990;47(5):465-473.
FREE FULL TEXT
56. US Preventive Services Task Force. Screening for depression. http://www.ahrq.gov/clinic/uspstf/uspsdepr.htm. Accessed January 23, 2008.57. Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff Syndrome. 2nd ed. Philadelphia, PA: FA Davis; 1989.58. Caine D, Halliday GM, Kril JJ, et al. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62(1):51-60.
FREE FULL TEXT
59. Harper C.. The incidence of Wernicke’s encephalopathy in Australia—a neuropathological study of 131 cases. J Neurol Neurosurg Psychiatry. 1983;46(7):593-598.
FREE FULL TEXT
60. Harper C, Fornes P, Duyckaerts C, et al. An international perspective on the prevalence of the Wernicke-Korsakoff syndrome. Metab Brain Dis. 1995;10(1):17-24.
PUBMED
61. Phillips SC. The threshold concentration of dietary ethanol necessary to produce toxic effects on hippocampal cells and synapses in the mouse. Exp Neurol. 1989;104(1):68-72.
PUBMED
62. Durand D, St Cyr JA, Gurevich N, Carlen PL. Ethanol-induced dendritic alterations in hippocampal granule cells. Brain Res. 1989;477(1-2):373-377.
PUBMED
63. Cadete-Leite A, Tavares MA, Pacheco MM, et al. Hippocampal mossy fiber CA2 synapses after chronic alcohol consumption and withdrawal. Alcohol. 1989;6(4):303-310.
PUBMED
64. Lundquist C, Alling C, Knoth R, Volk B. Intermittent ethanol exposure of adult rats: hippocampal cell loss after one month of treatment. Alcohol. 1995;30:737-748.
65. Bonthius DJ, West JR. Ethanol-induced neuronal loss in developing rats: increased brain damage with binge exposure. Alcohol Clin Exp Res. 1990;14(1):107-118.
PUBMED
66. Davis KM. WuJ-Y. Role of glutamatergic and GABAergic systems in alcoholism. J Biomed Sci. 2001;8(1):7-19.
PUBMED
67. Tsai G, Coyle JT. The role of glutamatergic neurotransmission in the pathophysiology of alcoholism. Annu Rev Med. 1998;49:173-184.
PUBMED
68. Kril JJ, Halliday GM. Brain shrinkage in alcoholics: a decade on and what have we learned? Prog Neurobiol. 1999;58(4):381-387.
PUBMED
69. Pfefferbaum A, Sullivan EV, Hedehus M, et al. In vivo detection and functional correlates of white matter microstructural disruption in chronic alcoholism. Alcohol Clin Exp Res. 2000;24(8):1214-1221.
PUBMED
70. Harper C, Dixon G, Sheedy D, Garrick T. Neuropathological alterations in alcoholic brains: studies arising from the New South Wales Tissue Resource Centre. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(6):951-961.
PUBMED
71. Ihara H, Berrios GE, London M. Group and case study of the dysexecutive syndrome in alcoholism without amnesia. J Neurol Neurosurg Psychiatry. 2000;68(6):731-737.
FREE FULL TEXT
72. Bendszus M, Weijers HG, Wiesbeck G, et al. SequentialMR imaging and proton MR spectroscopy in patients who underwent recent detoxification for chronic alcoholism: correlationwith clinical and neuropsychological data. AJNRAmJNeuroradiol. 2001;22(10):1926-1932.
FREE FULL TEXT
73. Gazdzinski S, Durazzo TC, Meyerhoff DJ. Temporal dynamics and determinants of whole brain tissue volume changes during recovery from alcohol dependence. Drug Alcohol Depend. 2005;78(3):263-273.
PUBMED
74. Bartsch AJ, Homola G, Biller A, et al. Manifestations of early brain recovery associated with abstinence from alcoholism. Brain. 2007;130(pt 1):36-47.
FREE FULL TEXT
75. Ruitenberg A, van Swieten JC, Witteman JCM, et al. Alcohol consumption and the risk of dementia: the Rotterdam Study. Lancet. 2002;359(9303):281-286.
PUBMED
76. Launer LJ, Feskens EJ, Kalmijn S, et al. Smoking, drinking, and thinking: the Zutphen Elderly Study. Am J Epidemiol. 1996;143(3):219-227.
FREE FULL TEXT
77. Galanis DJ, Joseph C, Masaki KH, et al. A longitudinal study of drinking and cognitive performance in elderly Japanese American men: the Honolulu Asia Aging Study. Am J Public Health. 2000;90(8):1254-1259.
FREE FULL TEXT
78. Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris). 1997;153(3):185-192.
PUBMED
79. Lemeshow S, Letenneur L, Dartigues JD, et al. Illustration of analysis taking into account complex survey considerations: the association between wine consumption and dementia in the PAQUID Study. Am J Epidemiol. 1998;148(3):298-306.
FREE FULL TEXT
80. Carmelli D, Swan GE, Reed T, et al. The effect of apolipoprotein E epsilon-4 in the relationships of smoking and drinking to cognitive function. Neuroepidemiology. 1999;18(3):125-133.
PUBMED
81. Dufouil C, Tzourio C, Brayne C, et al. Influence of apolipoprotein E genotype on the risk of cognitive deterioration in moderate drinkers and smokers. Epidemiology. 2000;11(3):280-284.
PUBMED
82. Truelsen T, Thudium D, Gronback M. Amount and type of alcohol and risk of dementia: the Copenhagen City Heart Study. Neurology. 2002;59(9):1313-1319.
FREE FULL TEXT
83. Mukamal KJ, Kuller LH, Fitzpatrick AL, et al. Prospective study of alcohol consumption and risk of dementia in older adults. JAMA. 2003;289(11):1405-1413.
FREE FULL TEXT
84. Brust JCM. Wine, flavonoids, and the "water of life." Neurology. 2002;59(9):1300-1301.
FREE FULL TEXT
85. Fillmore KM, Kerr WC, Stockwell T, et al. Moderate alcohol use and reduced mortality risk: systematic error in prospective studies. Addict Res Theory. 2006;14:101-132.
86. Folstein MF, Folstein SE, McHugh PR. A practical method for grading the cognitive state for the clinician. J Psychiatr Res. 1975;12(3):189-198.
PUBMED
87. Tangalos EG, Smith G, Ivnik RJ, et al. The Mini-Mental State Examination in general medical practice: clinical utility and acceptance. Mayo Clin Proc. 1996;71(9):829-837.
FREE FULL TEXT
88. Kukull WA, Larson EB, Teri L, et al. The Mini-Mental State Examination score and the clinical diagnosis of dementia. J Clin Epidemiol. 1994;47(9):1061-1067.
PUBMED
89. Katzman R, Rowe JR. Principles of Geriatric Neurology. Philadelphia, PA: Davis; 1992.90. Larson EB, Reifler BV, Sumi SM, et al. Diagnostic tests in the evaluation of dementia. Arch Intern Med. 1986;146(10):1917-1922.
FREE FULL TEXT
91. Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352(23):2379-2388.
PUBMED
92. Verghese J, LeValley A, Derby C, et al. Leisure activities and the risk of amnestic mild cognitive impairment in the elderly. Neurology. 2006;66(6):821-827.
FREE FULL TEXT
93. Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348(25):2508-2516.
PUBMED
94. Rundek T, Bennett DA. Cognitive leisure activities, but not watching TV, for future brain benefits. Neurology. 2006;66(6):794-795.
FREE FULL TEXT
95. Wang JYJ, Zhou DHD, Li J, et al. Leisure activity and risk of cognitive impairment: the Chongqing Aging Study. Neurology. 2006;66(6):911-913.
FREE FULL TEXT
96. Shumaker SA, Legault C, Coker LH. Behavior-based interventions to enhance cognitive functioning and independence in older adults. JAMA. 2006;296(23):2852-2854.
FREE FULL TEXT
97. Willis SL, Tennstedt SL, Marsiske M, et al. Long-term effects of cognitive training on everyday functional outcomes in older adults. JAMA. 2006;296(23):2805-2814.
FREE FULL TEXT
98. Price BH, Richardson EP Jr. The neurologic illness of Eugene O’Neill—a clinicopathological report. N Engl J Med. 2000;342(15):1126-1133.
PUBMED
99. Victor M, Adams RD, Mancall EL. A restricted form of cerebellar cortical degeneration occurring in alcoholic patients. Arch Neurol. 1959;1:577-688.
FREE FULL TEXT
100. Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565-598.
PUBMED
101. Pendery ML, Maltzman IM, West IJ. Controlled drinking by alcoholics: new findings and a reevaluation of a major affirmative study. Science. 1982;217(4555):169-175.
FREE FULL TEXT
102. Stern Y, Gurland B, Tatemichi TK, et al. Influence of education and occupation on the incidence of Alzheimer’s disease. JAMA. 1994;271(13):1004-1010.
FREE FULL TEXT
103. Snowdon DA, Kemper SI, Mortimer JA, et al. Linguistic ability in early life and cognitive function and Alzheimer’s disease in late life: findings from the nun study. JAMA. 1996;275(7):528-532.
FREE FULL TEXT
ARTICLES EN RAPPORT
Alcool, dépendance, impact social et sur la santé
Philippe Batel
JAMA. 2008;299:E1-E2.
Texte Complet
Neuropathie périphérique
Janet M. Torpy, Jennifer L. Kincaid, et Richard M. Glass
JAMA. 2008;299:1096.
Texte Complet
Cette semaine dans le JAMA-Français
JAMA. 2008;299:989.
Texte Complet
|