Antioxidants in Hypertension and Cardiovascular Disease
Abstract
Cardiovascular disease is characterized by enhanced oxidative stress in the vascular wall, heart, kidney, and brain. Epidemiological evidence suggests that antioxidants, including vitamins C and E, α-carotene, and β-carotene, may be therapeutic; however, interventional trials of antioxidants have provided mixed results, with some showing deleterious consequences. It is thus crucial that we consider the implications of trial design and execution, and further investigation of cellular pro-and antioxidant mechanisms is critical. Angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and mineralocorticoid receptor blockers reduce the generation of reactive oxygen species, in experimental models as well as in humans, and have demonstrated beneficial cardiovascular effects. Polyphenols and antioxidants contained in foods and beverages may also be cardioprotective. Recent studies suggest that the judicious development of antioxidant agents may provide an effective approach to quench oxidative stress in tissues and improve cardiovascular health.
Introduction
Oxidative stress is enhanced in hypertension, atherosclerosis, and other forms of cardiovascular disease and participates in the mechanisms of vascular injury. Reactive oxygen species (ROS) that induce oxidative stress include ·O2−, H2O2, · OH, HOCl and the reactive nitrogen species (RNS) nitric oxide (NO) and peroxynitrite (ONOO−). ROS and RNS are usually highly regulated and function as part of the intracellular signaling mechanisms of cells (1, 2). In hypertension, atherosclerosis, coronary artery disease (CAD), heart failure, diabetes, and other contexts of vascular damage, increased ROS production leads to endothelial dysfunction, enhanced contractility and growth of vascular smooth muscle cells (VSMCs), lipid peroxidation, inflammation, and increased deposition of extracellular matrix proteins. Markers of systemic oxidative stress are increased in both experimental and human hypertension (3, 4).
Treatment with antioxidants or superoxide dismutase mimetics may lower blood pressure and improve vascular structure and function in experimental and human hypertension (5, 6). Gene inactivation of ROS-generating enzymes (e.g., NOX1 or NOX2) lowers blood pressure in mouse models of acute angiotensin II–(AngII)-induced hypertension (1). Antioxidant reserve is reduced in tissues from hypertensive rodents and humans, as is evidenced by increased amounts of ROS. For these reasons, it has been argued that treatment with antioxidants could be beneficial in patients with cardiovascular disease, hypertension, atherosclerosis, CAD, or diabetes.
ROS can be produced by the actions of NADPH oxidase, uncoupled nitric oxide synthase (NOS), mitochondrial electron transport, xanthine oxidase, cyclooxygenase, lipoxygenase, heme oxygenase, and cytochrome P450 monooxygenase. Of these, NADPH oxidase, xanthine oxidase, uncoupled NOS, and mitochondrial electron transport are the major producers of ROS in the vascular wall. Reduced antioxidant capacity also promotes cellular oxidative stress and is implicated in cardiovascular and renal oxidative damage in hypertension. Superoxide dismutase, catalase, and glutathione peroxidase activities are reduced in hypertensive patients (7–9). Accordingly, antioxidant supplementation could have favorable therapeutic effects to reduce oxidative stress in cardiovascular disease (10, 11).
Molecular Targets of Reactive Oxygen Species in Vascular Cells
ROS function as mediators in many signaling pathways in association with redox-sensitive molecules that include transcription factors, protein tyrosine phosphatases, protein tyrosine kinases, mitogen-activated protein kinases, and ion channels (12). Transcription factors such as NF-κB, AP-1, and HIF-1 appear to be directly regulated by ROS (13), whereas other signaling molecules are indirectly regulated. Thus, the elevation of ROS to pathophysiological levels is associated with inflammatory responses, collagen deposition, and alterations in matrix metalloproteinase (MMP) activities; these responses, in turn, can lead to remodeling of the vasculature and uncoupling of NOS. Elevated ROS also decrease the bioavailability of NO, which is depleted upon reaction with superoxide anion to form ONOO−. ROS thus lead to a complex interplay of chemical and biomolecular events that contribute to cardiovascular disease.
Oxidative Stress and Clinical Hypertension
Although studies in humans have not been as convincing as those in experimental models, there is evidence that oxidative stress is increased in patients with essential hypertension, renovascular hypertension, malignant hypertension, salt-sensitive hypertension, cyclosporine-induced hypertension, and preeclampsia (14–16). These findings are based, in large part, on increased levels of plasma thiobarbituric acid–reactive substances and 8-epi-isoprostanes, biomarkers of lipid peroxidation and oxidative stress. The generation of superoxide anion by polymorphonuclear leukocytes and platelets, which also participate in vascular oxidative stress and inflammation, is also elevated in hypertensive patients.
Hypertensive patients exhibit a significantly higher production of plasma H2O2 than normotensive subjects (17). Additionally, normotensive subjects with a family history of hypertension manifest greater H2O2 production than do blood pressure–matched normotensives without a family history of hypertension, suggesting that there may be a genetic component that leads to elevated production of H2O2 (18). Plasma levels of asymmetric dimethylarginine (ADMA), an inhibitor of endothelial NOS (eNOS), and 13-hydroxyoctadecadienoic acid, a marker of ROS production, are elevated in patients with hypertension, and microvessels from patients with the highest levels of these markers are least responsive in terms of NO-induced relaxation (19).
ROS production is increased in VSMCs from resistance arteries of hypertensive patients and is associated with upregulation of vascular NADPH oxidase (20). Indeed, the importance of NADPH oxidase in contributing to oxidative stress and human cardiovascular disease is reflected in a number of polymorphisms in NADPH oxidase that occur in atherosclerosis and hypertension (21). Some isoforms of NADPH oxidase may be more relevant than others, which is an important area of investigation into the pathogenesis of hypertension. For example, patients with chronic granulomatous disease who are deficient in the prototypical NOX2 isoform (see below) do not display significant alterations in blood pressure.
In addition to mechanisms that generate excess ROS, mechanisms that interfere with antioxidant defense are also at play in hypertensive patients. Diminished antioxidant defense can stem from deficiencies in superoxide dismutase, glutathione peroxidase, or catalase (22). Moreover, superoxide dismutase activity correlates inversely with blood pressure in patients with hypertension (22, 23). Accordingly, antioxidant vitamins reduce blood pressure and arterial stiffness in patients with diabetes but seem to have no effect in postmenopausal women, in healthy subjects, or in the prevention of preeclampsia (23–25).
Atherosclerosis
Oxidative stress plays an important role in atherogenesis (Figure 1) by promoting the oxidation of lipids and proteins in the vascular wall and the proliferation and migration of smooth muscle cells to the intima (26, 27); atherosclerosis lesions and endothelial dysfunction have both been linked to excessive ROS generation. Significantly, common cardiovascular risk factors that promote atherosclerosis (e.g., hypercholesterolemia, diabetes mellitus, hypertension, smoking, age, and nitrate intolerance) increase ROS production.
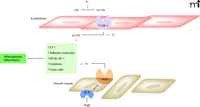
Atherogenesis exemplifies mechanisms of ROS signaling and damage in the cardiovascular system. Superoxide anion (·O2–) is depicted in promoting the oxidation of lipids and related effects on adhesion, migration, and signaling (see text for details). AngII-mediated effects, including upregulation of NADPH oxidase (NOX) and LOX-1, are also indicated.
The renin-angiotensin system (RAS) is also an important factor in redox-sensitive atherogenesis. For example, the AT1 receptor of angiotensin AngII is upregulated in atheromatous plaques (28). In fact, AngII elicits multiple responses involved in redox physiology and inflammation, including: upregulation of LOX-1, the human endothelial receptor for oxidized low-density lipoprotein (LDL) (29); uptake of oxidized LDL by macrophages (30); and progression of atherosclerosis in apoE-deficient mice (31). MMPs are stimulated by AngII, which contributes to the progression of plaque formation and instability (32, 33). Vascular oxidant stress also enhances progression and angiogenesis of experimental atheroma (34). Accordingly, the blockade of the RAS with inhibitors of angiotensin converting enzyme (ACE) or AT1 receptor blockers promotes vascular protection (35).
The overexpression of vascular smooth muscle cell NADPH oxidase (i.e., the p22phox subunit) in mice is associated with enlarged arterial lesions (1). Elevated levels of hydrogen peroxide and vascular endothelial growth factor (VEGF) are also associated with overexpression of NADPH oxidase, likely reflecting the increased expression of HIF-1α induced by oxidative stress; accordingly, the development of lesions in this mouse model is associated with neointimal angiogenesis. Neovascularization has been well described in atheroma and is considered to play an important role in the progression of atherosclerosis and in plaque instability (35). In particular, oxidative stress is envisaged to trigger an angiogenic switch leading to angiogenesis and atheroma progression, and in this regard, it is significant that NADPH oxidase (i.e., the p47phox subunit) is required for plaque progression in hypercholesterolemic mice (36). The predominant ROS related to this requirement is H2O2, which thus appears to be a component of the angiogenic switch.
ROS generation by NADPH oxidase has been demonstrated in human atherectomy specimens (37). Particular catalytic subunits of NADPH oxidase isoforms (i.e., gp91phox, Nox 1, and Nox4) may contribute to increased oxidative stress in human coronary atherosclerosis in a cell-specific manner (38). In coronary artery segments from explanted human hearts, ROS formation occurs throughout the intima, media, and adventitia of nonatherosclerotic coronary arteries. In atherosclerotic arteries, ROS generation is increased in the plaque shoulder, which is rich in macrophages and α-actin–positive cells. The p22phox subunit of NADPH oxidase colocalizes with gp91phox mainly in macrophages, whereas Nox4 is found in nonphagocytic vascular (α-actin–positive) cells. Levels of gp91phox- and p22phox-encoding mRNAs correlate with the severity of atherosclerosis. NOX1 expression is low both in human coronary arteries and isolated vascular cells.
NADPH oxidase is not the only source of ROS that can influence progression of atherosclerosis. Xanthine oxidase, myeloperoxidase, and eNOS have also been implicated (39). Recently, the role of mitochondrial dysfunction, which can be caused by AngII, has been pinpointed in endothelial dysfunction and atherosclerosis. Mitochondria are both sources and targets of ROS, and there is growing evidence that mitochondrial dysfunction may be involved in the formation of vascular lesions (40). In atherosclerotic lesions of apoE-null mice, aldose reductase occurs in macrophage-rich regions and in proportion to lesion progression (41). This occurrence likely reflects a protective (antioxidant) role for aldose reductase, as pharmacological inhibition or genetic ablation of the enzyme promotes lesion formation. Protective functions may also be provided by superoxide dismutase and catalase. Other enzymes, such as heme oxygenase (HO)-1, which is the rate-limiting enzyme in the catabolism of heme, also appear to be protective. HO-1 activity, which is essential to the generation of the antioxidant biliverdin, iron, and carbon monoxide, counters atherogenesis with both antioxidant and anti-inflammatory functions. HO-1–deficient macrophages in null mice are associated with higher levels of ROS and proinflammatory cytokines such as MCP-1 and IL-6 (42). They also exhibit enhanced foam cell formation when treated with oxidized LDL.
The oxidation of LDL species appears to have evolved as an important indicator of cellular damage for activating receptor-mediated pathways. The lectin-like LOX-1 receptor is responsible for the binding and uptake of oxidized LDL in endothelial cells and is upregulated under proatherogenic conditions such as diabetes, hypertension, and dyslipidemia. Aortic atherosclerosis is greatly reduced when LOX-1 activity is knocked out in mice containing an LDL receptor-(LDLR)-null background; the knockout of LOX-1 in these mice results in the expression of anti-inflammatory signals (43).
Other mechanisms that can promote atherosclerosis have been established, although they are perhaps not as widely recognized. The proteasome, for example, contributes to cellular protection against oxidative stress, and chronic proteasome inhibition appears to promote coronary artery oxidative stress and early atherosclerosis in pigs (44). Markers of vascular senescence have also been linked to human atheroma, including senescence-associated α-galactosidase (SAαG) and p16 and p21, which are cyclin-dependent kinase inhibitors (CDKIs) not found in healthy vessels (45). Plaques and fibrous cap smooth muscle also have shorter telomeres, such that telomere shortening correlates with severity of atherosclerosis. Vascular senescence is mediated by changes in cyclins D/E, p16, p21, and retinoblastoma protein (pRB), and is associated with low telomerase activity. Indeed, telomerase expression alone rescues plaque vascular cell senescence, despite telomere shortening, and normalizes the CDKI/pRB changes. For this reason, telomere damage may be secondary to oxidant stress. Oxidants induce premature senescence in vitro, with accelerated telomere shortening and reduced telomerase activity. These data bring together aging, telomerase deficiency and shortened telomeres, oxidative stress, and exaggerated atherogenesis via oxidative stress–induced DNA damage.
Antioxidant Therapy and Cardiovascular Disease
There has been great interest in targeting ROS to treat or prevent cardiovascular disease, including atherosclerosis and hypertension. Attempts to target ROS have been based on increasing antioxidant bioavailability (through diet or supplements) and reducing ROS generation (through inhibition of superoxide-generating enzymes). The ability of antioxidants to treat conditions associated with oxidative stress is supported by experimental, observational, and epidemiological studies in humans (46–62), although the findings have not been consistent, with most large trials demonstrating negative effects of antioxidants on cardiovascular outcomes. A recent study investigating the effects of vitamins C and E on the development of hypertension in pregnancy, for example, failed to show any benefit of antioxidant vitamins in hypertension (58). In contrast, smaller clinical studies, which were well controlled and which investigated effects of antioxidants specifically on blood pressure, have demonstrated significant beneficial effects (59–62). Based on current data, it is recommended that the general population should consume a balanced diet (e.g., the DASH diet) with emphasis on antioxidant-rich fruits and vegetables and whole grains (63–65). Table 1 indicates components of an antioxidant-rich diet.
Antioxidants and Dietary Sources
Vitamin E
Epidemiological studies that indicated an inverse relationship between the intake of fruits and vegetables and the appearance of cardiovascular disease were initially taken to reflect the benefit of antioxidant vitamins in the diet. Two prospective studies that evaluated the impact of vitamin E were the Nurses’ Health Study of 87,245 female nurses for eight years (66) and the Male Health Professionals’ Study of 39,910 male health professionals for four years (67). In both studies, the intake of more than 100 IU of vitamin E per day for more than two years was associated with reduced risk of CAD. A Finnish study of 5000 men and women found a statistically significant cardiovascular benefit with vitamin E supplementation for women but not for men (68). In the Women’s Health Study (69), 39,876 apparently healthy US women over forty-five years of age were randomly assigned, using a 2x2 factorial design, to receive vitamin E or placebo and aspirin or placebo; follow-ups occurred at an average of 10.1 years, with natural-source vitamin E received at a dose of 600 IU on alternate days. This dose of vitamin E provided no benefit for major cardiovascular events or cancer, did not affect total mortality, but decreased cardiovascular mortality in healthy women.
β-Carotene
The Nurses’ Health Study did not find any benefit from β-carotene after taking into account the intake of vitamins C and E (66). The follow-up Male Health Professionals’ Study reported a beneficial effect of β-carotene, even after adjusting for intake of vitamin C and E, for smokers but not for nonsmokers (67). The Finnish study was unable to detect any reduction in cardiovascular risk that could be attributed specifically to β-carotene (68).
Primary and Secondary Prevention Randomized Clinical Trials
The Cambridge Heart Antioxidant Study (CHAOS) was a randomized, placebo-controlled, double-blind trial of 2,002 patients with CAD (proven by coronary angiography) who received up to 800 IU of vitamin E versus placebo for less than three years (53). There was a dramatic reduction in fatal myocardial infarction; however, there were problems of design and randomization, and many patients died in the vitamin E group. The α-Tocopherol, β-Carotene Cancer (ATBC) Prevention Study was also a double-blind, placebo-controlled, randomized clinical trial of primary prevention of lung and other cancers carried out in Finland on male smokers for up to eight years (70). There was no effect on cancer. There were slight benefits for incident angina, but no reduction of cardiovascular deaths and an increase in hemorrhagic stroke; the dose of vitamin E (50 mg per day) may have been too small. A meta-analysis of interventional trials suggests low doses of vitamin E may be ineffective whereas higher doses could in fact be harmful (71).
The ATBC trial found increased mortality in the subjects taking β-carotene, with trends to increased lung cancer and ischemic heart disease (70). In the β-Carotene and Retinol Efficacy Trial (CARET), 18,314 smokers, former smokers, and asbestos workers were treated with β-carotene and retinol or placebo for up to four years (71). The trial had to be stopped early because of increased incidence of lung cancer in the β-carotene and retinol group. The Physicians’ Health Study I (PHS I) included 22,071 male physicians for a mean of twelve years (72) and concluded that there was neither a beneficial nor a deleterious effect of 50 mg per day of β-carotene on cancer incidence, CAD, or mortality. The Physicians’ Health Study II was performed on 7,641 volunteers (41%) from the PHS I who retained their original β-carotene treatment assignment and 7000 new physicians who were randomized in a 2x2x2x2 factorial design with mean follow-up of eight years (73). No beneficial effect on major cardiovascular events, total myocardial infarction, total stroke, and cardiovascular mortality could be discerned from this study.
Secondary prevention trials with a combination of vitamin E (600 mg), vitamin C (250 mg), and β-carotene (20 mg) in the Heart Protection Trial in the UK (20,536 subjects) demonstrated no cardiovascular benefits (58). The Women’s Antioxidant Cardiovascular Study (WACS) in the US recruited 8,171 female health professionals at increased risk in a 2×2×2 factorial design (74). The participants were forty years of age or older with a prior history of cardiovascular disease or three or more risk factors and were followed for an average of 9.4 years, but no beneficial cardiovascular effect was observed. In HOPE-TOO (75), 4,732 of the initial HOPE participants agreed to extended observation and were evaluated every six months. Of these, 3,994 (i.e., 65.3% of those surviving) agreed to continue in the study and were followed for a median of seven years. Long-term vitamin E supplementation did not prevent cancer or major cardiovascular events and could increase the risk for heart failure.
Prevention Trials of Antioxidant Protocols: Assessing Negative Results
Possible explanations for the failure of primary and secondary prevention trials to reveal beneficial antioxidant effects on cardiovascular risk are many. Dosing and effective duration of exposure may have been problematic, and interactions between agents have not been fully investigated. Cellular compartmentalization of antioxidants may also be a challenge, such that potential therapeutics do not reach appropriate targets. Vitamin E, for example, concentrates in lipoproteins and may thus not reach cytoplasmic ROS. [On the other hand, an agent that targets the mitochondrion (i.e., mitoTEM-PO) may buffer mitochondrial superoxide generation and attenuate Ang II–induced hypertension (76).] Additionally, vitamins E and C may, subsequent to metabolic conversion, act as pro-oxidants, thereby undermining their therapeutic potential. Furthermore, patients who were recruited may have had advanced cardiovascular disease, as a result of oxidant injury, and may not have been amenable to intervention. Many patients were taking aspirin, which has intrinsic antioxidant properties (77), defeating observational accuracy. Finally, it has never been proven that the trial subjects in these different studies indeed had increased oxidative stress in their cardiovascular system, although this seems likely, and in none of the trials discussed here was blood pressure a primary end point.
Strategies to Reduce ROS Generation
Some of the beneficial effects of classical antihypertensive agents, such as β-adrenergic blockers (particularly carvedilol, which is endowed with antioxidant properties), ACE inhibitors, AT1 receptor antagonists, mineralocorticoid receptor antagonists, and Ca2+ channel blockers, may be partially mediated by decreasing vascular oxidative stress (78–82). These effects have been attributed to direct inhibition of NADPH oxidase activity and to intrinsic antioxidant properties of the drugs. Other drugs that reduce oxidative stress include HMG-CoA reductase inhibitors (statins), allopurinol, PPARγ activators, and aspirin (Table 2). Nonpharmacological factors such as diet (Mediterranean, DASH) and regular exercise are critical. In experimental models of hypertension and in human patients with CAD, exercise reduced vascular NADPH oxidase activity and ROS production, ameliorated vascular injury, and reduced blood pressure (83).
Approaches to Reduce Oxidant Stress
Theoretically, agents that reduce oxidant formation should be more efficacious than nonspecific, inefficient antioxidant vitamin scavengers. This is based on experimental evidence demonstrating that inhibition of NADPH oxidase–mediated superoxide generation (through pharmacological and gene-targeted strategies) leads to regression of vascular remodeling, improved endothelial function, lowering of blood pressure, and prevention of atherosclerosis. Strategies to increase antioxidant capacity without the use of exogenous antioxidants are possible by using drugs that selectively inhibit MRP1 to prevent cellular glutathione loss and thereby protect against oxidative damage, endothelial dysfunction, and hypertension (84). Another interesting approach is targeting glucose-6-phosphate dehydrogenase (G6PD), which is a source of NOX substrate (i.e., NADPH) (85). Inhibition of G6PD has been shown to ameliorate development of pulmonary hypertension, possibly through decreased oxidative stress. To date, only investigational G6PD inhibitors are available.
Resveratrol, Dark Chocolate, and Other Polyphenols
Epidemiological evidence has suggested that polyphenols from fruits, vegetables, green and black tea, and some wines can decrease cardiovascular risk. Green tea polyphenols, for example, improve endothelial function and insulin sensitivity, reduce blood pressure, and protect against myocardial ischemia/reperfusion injury (86). Polyphenols of interest may include resveratrol, naringenin, quercetin, and catechins. Resveratrol, perhaps the best-known of these, appears to exert its effects in part by stimulating endothelial SIRT1, which regulates endothelium-dependent vasodilation (87). Resveratrol also appears to affect nuclear factor-E2-related factor-2 (Nrf2) activity (88).
Flavonoid-rich dark chocolate compares favorably with flavonoid-free white chocolate regarding effects on blood pressure and insulin sensitivity in hypertensive patients (90). Recently, a population-based prospective study of middle-aged and elderly women demonstrated that the regular and moderate consumption of chocolate (i.e., less than one serving per day) engendered a lower incidence of heart failure, hospitalization, and death (91).
Conclusions
Oxidative stress in hypertension and other cardiovascular conditions contributes to vascular injury by promoting VSMC growth, endothelial dysfunction, inflammation, increased vascular tone, and MMP activation. These processes lead to altered vascular contractility and structural remodeling, characteristic features of vessels in hypertension and other forms of cardiovascular disease. Foods such as dark chocolate, along with flavonoids in red wine and inhibitors of the renin-angiogensin-aldosterone system, suggest that antioxidant treatments may be vasculoprotective. New knowledge of mechanisms involved in oxidant stress in tissues, ROS production, and the fate of antioxidants after their administration may lead to progress in improved outcomes and refined therapies for hypertension and other forms of cardiovascular disease.
Acknowledgments
Studies from the author’s laboratory were supported by funds from the Canadian Institutes of Health Research (CIHR) [Grants 37917, 82790] and a Canada Research Chair (CRC) from the CIHR/ Government of Canada CRC Program and by the Canada Fund for Innovation.
- Copyright © 2010
References

Ernesto L. Schiffrin, MD, PhD, is Professor and Vice-Chair of Research of the Department of Medicine at McGill University and holds a Canada Research Chair at the Lady Davis Institute of the Jewish General Hospital. E-mail ernesto.schiffrin{at}mcgill.ca; fax 514-340-7539.

