Matters of the Heart: The Case of TNFα-Targeting Drugs
- Fabio Cacciapaglia1,4,
- Pierantonio Menna2,4,
- Luca Navarini3,4,
- Antonella Afeltra3,4,
- Emanuela Salvatorelli2,4 and
- Giorgio Minotti2,4
- 1 Internal Medicine and Rheumatology, “N. Melli” Hospital, San Pietro Vernotico, Brindisi
- 2 Drug Sciences and Clinical Pharmacology, University Campus Bio-Medico of Rome
- 3 Clinical Medicine and Rheumatology, University Campus Bio-Medico of Rome
- 4 Center for Integrated Research and Alberto Sordi Foundation for Research on Aging, University Campus Bio-Medico of Rome, Italy
Drugs targeted at tumor necrosis factor–alpha (TNFα) have paved new avenues for treating chronic inflammatory diseases such as rheumatoid arthritis and many others. Anti-TNFα drugs have been also considered as therapeutics to treat heart failure or other cardiovascular events that are caused or accompanied by high circulating levels of TNFα. Surprisingly, however, anti-TNFα therapies caused both beneficial and detrimental cardiovascular effects. Here, we suggest that the disappointing cardiovascular outcome of pharmacological interventions with TNFα-targeted drugs could be reconciled with the Janus-like behavior of TNFα, a molecule that conveys both negative and positive stimuli to the heart.
Many drugs developed and approved to treat noncardiovascular diseases are known to cause cardiovascular effects that range from arrhythmias to blood pressure disorders, ischemia, heart failure (HF), thromboembolism, or combinations thereof. Drugs labeled as possibly cardiotoxic belong to such diverse classes as antineoplastics, nonsteroidal anti-inflammatory drugs, antihistamines, psychoactive drugs, antibiotics, and antiretrovirals (1). This viewpoint paper aims at familiarizing the reader with a potential new family of cardiotoxic drugs, the tumor necrosis factor–alpha (TNFα)-targeted drugs.
In comparison to noncardiovascular drugs known or suspected to cause cardiotoxicity, TNFα-targeted drugs provide a case for more complexity and ambiguity. Complexity exists because TNFα-targeted drugs were developed to treat patients with inflammatory diseases known to be promoted or sustained by TNFα. In such patients, however, TNFα-targeting drugs caused no effect on cardiovascular events associated with systemic inflammation, or they caused reduced incidence in some studies or a trend toward increased incidence in other studies. Ambiguity exists because TNFα-targeted drugs were repurposed as cardiovascular drugs. In fact, patients who had not had rheumatic disease but were affected by heart failure (HF) have long been known to carry high serum concentrations of TNFα, which suggests that drugs able to antagonize TNFα could be of benefit to these patients (2). Unfortunately, studies that probed “anti-TNFα” drugs in nonrheumatic patients showed clinical worsening or no improvement of HF.
Complexity and ambiguity might be reconciled if we take into consideration that patients who apparently share TNFα as a marker of disease may have differing underlying disease etiologies. Although reasonable and clinically sound, this conclusion would neglect the supposition that the subtleties of TNFα concentration and localization may exert influences that add complexity and ambiguity to the conclusions drawn from research on this cytokine. Therefore, defining the risk:benefit of anti-TNFα drugs in cardiovascular settings requires a brief reppraisal of what TNFα does in healthy or diseased conditions.
TNFα is a pleiotropic cytokine that participates in many aspects of the innate immune system, regulates the responses of immune cells, and thus stimulates the acute-phase reaction that occurs as a response to inflammation (3). Infectious or inflammatory stimuli induce the release of large amounts of TNFα into the systemic circulation, which allows TNFα to modulate the expression of a constellation of factors involved in the inflammatory reaction [interleukin-1 (IL-1), IL-6, platelet-derived growth factor (PDGF), tumor growth factor–beta (TGFβ), eicosanoids, platelet-activating factor (PAF), and adrenaline]. TNFα activates and promotes leucostasis by inducing increased expression of adhesion molecules at sites of inflammation. These effects are seen at TNFα plasma concentrations below 10−9 M and denote the physiological role of TNFα (4). At higher plasma concentrations, however, TNFα induces less benign endocrine- and exocrine-like effects observed in endotoxemia and shock (i.e., metabolic wasting, microvascular clotting, and hypotension). TNFα also causes diverging effects on tissue integrity and turnover. On the one hand, TNFα may assist tissue repair by stimulating fibroblast and mesenchymal cell proliferation, either directly or through inducing expression of growth factors; on the other hand, TNFα causes dysfunction or apoptosis of endothelial cells, increases the expression of collagenases and proteases, and thus contributes to tissue damage. Supraphysiological levels of reactive oxygen species (ROS) and cyclooxygenase metabolites of arachidonic acid (prostaglandins) usually accompany the untoward effects of TNFα (5, 6). Thus, overproduction of TNFα appears to cause or accompany autoimmunity and inflammation and has been implicated in rheumatoid arthritis, psoriasis, psoriatic arthritis, ankylosing spondylitis, inflammatory bowel diseases, vasculitis, multiple sclerosis, and sarcoidosis.
In the era of DNA recombination and protein engineering, anti-TNFα drugs presented as an unprecedented option to treat inflammatory diseases caused or sustained by TNFα. Currently approved anti-TNFα drugs can be subgrouped as monoclonal antibodies (i.e., infliximab, adalimumab, and golimumab), chemically modified Fab’ fragments of TNFα-specific monoclonal antibodies (i.e., certolizumab-pegol), or fusion proteins that carry the ligand-binding moiety of TNFαreceptor 2 (i.e., etanercept) (Table 1). TNFα-targeting drugs can be used as single agents in patients who failed to respond to disease-modifying antirheumatic drugs (DMARD) like methotrexate, leflunomide, or sulphasalazine, but there are regimens that combine anti-TNFα drugs with DMARDs and show higher efficacy than anti-TNFα drugs alone. Anti-TNFα drugs are only available in parenteral formulations; attempts to improve patient compliance by designing orally bioavailable drugs are still in their infancy.
Anti-TNFα Drugs and Approved Indications
The clinical development of other anti-TNFα drugs has been less favorable. Testing of CDP 571, a humanized antibody to TNFα, was halted after a disappointing Phase II study of patients with Crohn’s disease, whereas the clinical development of onercept, a recombinant, unmodified, fully humanized, soluble type 1 TNF receptor (p55), was stopped because of its lack of efficacy in Crohn’s disease and its unfavorable risk:benefit in patients with moderate-to-severe psoriasis. Nonetheless, anti-TNFα drugs introduced long-awaited improvements in the clinical management of chronic inflammatory diseases and expanded a pharmacological repertoire that for too long had remained bound to the relatively nonspecific DMARDs. To some extent, anti-TNFα drugs could be compared to the so-called targeted drugs that revolutionized pharmacological approaches to anticancer therapy. That is, in oncologic settings, targeted drugs were developed to overcome limitations of nonspecific cytostatics and cytotoxics and specifically to inhibit receptors, receptor-associated kinases, intracellular kinases, or other enzymes that were thought to be crucial to the growth and survival of cancer cells but not of normal cells (7).
Cardiovascular events represent the main cause of death in patients with rheumatoid arthritis (RA). These RA patients are at a twofold greater risk for myocardial infarction (MI) and stroke, which jumps to nearly threefold in patients who have had the disease for ten years or more (8, 9). The risk of cardiovascular disease and mortality is largely independent of traditional risk factors (such as obesity or smoking) but relates to chronic inflammation and consequent accelerated atherosclerosis (9, 10). Anti-TNFα drugs were therefore anticipated to protect RA patients from cardiovascular events, but unfortunately this was not the case. Studies that looked at the incidence of any cardiovascular event showed that the effects of anti-TNFα drugs varied from reducing the risk to lacking any measurable influence (11). Regarding MI (whether primary or recurrent), anti-TNFα drugs were reported to lack an effect, to reduce the risk, or to increase it (11). And finally, studies that looked at HF as the major contributor to cardiovascular mortality in RA patients showed a very disappointing blending of reduced risk, no effect, or increased risk (11).
In the late 1990s, preclinical studies showed that TNFα inhibition could protect against cardiac dysfunction (12). These observations, and the aforesaid correlation of HF severity with the circulating amounts of TNFα, paved the road to exploratory clinical studies and placebo-controlled phase II trials that probed etanercept in patients with New York Heart Association (NYHA) class III or IV HF (Box 1). All such studies were quite consistent in demonstrating improvements in left ventricular function and remodeling and clinical composite scores (13, 14). Etanercept also caused a remarkable improvement of endothelium-dependent vasodilation, which was consistent with a possible role of TNFα in inducing endothelial dysfunction in HF patients (15, 16). Everything looked promising and well set to move toward larger studies, but once again ambiguity and complexity surfaced. In the phase IIB study of anti-TNFα Therapy Against Chronic Heart failure (ATTACH), 150 patients with NYHA class III-IV were randomized to receive placebo or low- or high-dose infliximab. Left ventricular ejection fraction was only transiently improved by infliximab; at a longer follow-up, both infliximab groups showed a trend toward increasing mortality or hospitalization for HF (17, 18). Infliximab-associated deterioration of cardiac function occurred in the presence of diminished serum levels of acute-phase proteins (i.e., C-reactive protein and interleukin-6) (Figure 1). The unexpected results of ATTACH could only suggest a contraindication to using infliximab in patients with HF.
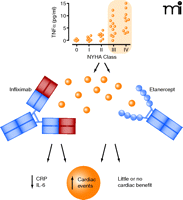
The graph in the upper section shows increasing circulating levels of TNFα in HF patients as a function of disease severity (NYHA class). The central section represents NYHA class III or IV patients that were recruited in trials that probed infliximab or etanercept. In the bottom section, infliximab decreased circulating levels of acute phase proteins (IL-6, CRP) but caused increased cardiac events, while etanercept lacked effects or caused increased cardiac events. Solid circles indicate circulating TNFα. NYHA, New York Heart Association; IL-6, interleukin 6; CRP, C–reactive protein. Based on (2, 17–19).

ATTACH was not the only study to uncover deleterious effects of anti-TNFα drugs (Figure 1). Two larger studies, RENAISSANCE (Randomised Etanercept North American Strategy to Study Antagonism of Cytokines) and RECOVER (Research into Etanercept Cytokine Antagonism in Ventricular Dysfunction), were terminated because of a lack of efficacy or a true negative result of low- or high-dose etanercept in patients with NYHA class III-IV. Analysis of RENEWAL, which merged the results from RENAISSANCE and RECOVER, showed that in RECOVER, high-dose etanercept demonstrates only slightly better results than low-dose etanercept, whereas in RENAISSANCE, the risk of death or hospitalization for HF is increased at both low- and high-dose etanercept (16, 19). These results led to contraindicate etanercept in nonrheumatic patients with HF and raised caution against using etanercept in RA patients with HF.
The diverging effects of anti-TNFα drugs on cardiovascular events in RA patients could at least in part be attributed to study-to-study differences in patient selection and evaluation (e.g., retrospective versus prospective studies, data retrieval from national registries or insurance records, absence or presence of preexisting comorbidities, timing and length of anti-TNFα therapy, monotherapy versus combination with DMARDs, and type of DMARD). Nonetheless, it is quite evident that TNFα dysregulation and systemic inflammation by no means anticipate that anti-TNFα drugs are salutary for the heart. This concept is echoed in the studies of non-rheumatic patients with HF: high TNFα concentrations do not always translate into a cardioprotective effect from anti-TNFα drugs, regardless of whether these drugs also decrease the serum levels of other reactive proteins. We have described that in many RA or nonrheumatic patients, anti-TNFα drugs actually caused more damage to the heart, particularly in the presence of HF. So, what is wrong with anti-TNFα drugs? We suggest that the experience gained with anti-TNFα drugs calls for revisiting the axiom “TNFαsynonymous to cardiac dysfunction”: this should be tempered and reshaped to incorporate appreciation of both detrimental and beneficial effects of TNFα. The unexpectedly negative effects of anti-TNFα drugs on cardiac function would accommodate the dual behavior of TNFα.
There are several proposed mechanisms that attest to the dual role of TNFα in the heart: 1) activation or increased expression of proapoptotic proteins like caspases and Bax (20, 21) but also increased transcription of genes related to cellular survival and proliferation (22, 23), and 2) activation of apoptosis signal–regulating kinase 1 but also of antiapoptotic Btk nonreceptor tyrosine kinases (24). The balance of cellular life versus death is governed by two distinct receptors, TNFα receptor 1 (TNFR1, also referred to as p55) and TNFα receptor 2 (TNFR2 or p75). The two receptors share significant sequence similarity in their extracellular domains but not in their cytoplasmic domains, which suggests that activation of one receptor or the other could result in different downstream effects. Accordingly, TNFR1 has been reported to signal apoptosis and fibrosis or remodeling, whereas TNFR2 has been reported to signal survival, repair, and angiogenesis (“cell survival receptor”) (25). Such opposing functions of TNFR1 and TNFR2 have been proposed to explain improved versus exacerbated post-infarction remodeling in TNFR1−/− or TNFR2−/− mice, respectively (26, 27), or mitigation versus aggravation of HF in TNFR1−/− or TNFR2−/− mice (28), respectively. In cardiomyocytes, TNRF1 is expressed more abundantly than TNRF2, which, in principle, would only suggest bad news for the heart (24); however, TNFα shows higher affinity for TNFR2, from which it may eventually “pass” to TNFR1 (29). Furthermore, there is compelling evidence that both TNFR1 and TNFR2 might be required to protect isolated cardiomyocytes or whole-heart preparations against hypoxia and reoxygenation or ischemia and reperfusion, as if TNFR1 and TNFR2 shared one or more cytoprotective repertoires (30, 31). TNFα receptor-associated factor 2 (TRAF2), a scaffolding protein that is common to TNFR1 and TNFR2, propagates cardiac survival effects in experimental models of ischemia-reperfusion injury (32). The binding interface of TRAF2 and other scaffolding proteins for TNFR1 is being investigated in depth (33). These studies will further clarify how TNFR1, previously reported to signal apoptosis, may assist TNFR2 in signaling survival and resistance to stressor agents.
The aforesaid beneficial effects of TNFα were seen in genetically manipulated mice that express different amounts of TNFR1 or TNFR2; however, similar effects had been seen in wild-type cells or in animals exposed to exogenous TNFα, particularly when low-dose TNFα was used to treat cardiomyocytes or to perfuse (i.e., precondition) isolated heart prior to subjecting it to ischemia-reperfusion (23, 34). High-dose TNFα did, in fact, suppress contractility in working isolated heart, or it made the heart more vulnerable by subsequent ischemia-reperfusion (35). Such a concentration dependence of the effects of exogenous TNFα is not too different from that observed with endogenous TNFα: in wild-type mice, ischemia-reperfusion evoked protective synthesis and release of TNFα from cardiomyocytes whereas mice that over-expressed TNFα exhibit accelerated ventricular inflammation and dysfunction (23, 36).
On balance, there seems to be a window of opportunity for endogenous or exogenous TNFα to convey cardioprotective signals, particularly when the concentration of either source of TNFα is low enough to preferentially activate TNFR2 or to drive the TNFR1-TNFR2 couple toward cytoprotective processes only. How these mechanisms precisely work is uncertain. For example, increased ROS formation and NF-κB activation were reported to transduce TNFα-mediated stimulation into both cardiac protection and injury (37–39). Nonetheless, linking “some TNFα” (i.e., low concentrations of TNFα) with cardioprotection twists our perception of TNFα antagonism in RA or nonrheumatic patients: it raises the possibility that intercepting TNFα would leave cardiac dysfunction unchanged or more aggressive.
The cardioprotective role of low concentrations of TNFα was inferred by comparing data from disparate experimental models of ischemia-reperfusion or HF. Can we next assume that “some TNFα” struggles to protect the heart against high TNFα concentrations that occur in patients with cardiac disease or are at risk for it? In other words, can we consider low TNFα to offer preconditioning benefits against high TNFα? The observation that anti-TNFαdrugs did not always diminish but actually increased the incidence of cardiac events might in fact be explained by assuming that they intercepted those discrete amounts of TNFα that heralded cardiomyocyte preconditioning and survival to high concentrations of TNFα.
Testing this hypothesis requires experimental and conceptual considerations. On experimental grounds, there is a need for unambiguous models that explore “low TNFα” against “high TNFα.” We have found that TNFα causes concentration-dependent death of isolated cardiomyocytes; however, pre-exposing cardiomyocytes to nontoxic concentrations of TNFα (one-sixth of its IC50 value) made them resistant to a rechallenging with high concentrations of TNFα (40). TNFα preconditioning offered a net survival effect over a broad range of otherwise toxic concentrations of TNFα and peaked at TNFα concentrations ~20 times higher than its IC50 value in unpreconditioned cells. Interestingly, low TNFα could also protect cardiomyocytes that were subsequently challenged with bolus H2O2 or etoposide to cause oxidative stress or apoptosis, respectively (41); however, TNFα was much better at protecting against etoposide rather than H2O2 (survival effect at concentrations ~ 40 times higher than or equal to respective IC50 values in unpreconditioned cells, respectively) (42). On balance, the effect of low TNFα against high TNFα was much similar to that of low TNFα against etoposide, which supports a prevailing role of apoptosis in TNFα cardiotoxicity.
The untoward cardiac effects of anti-TNFα drugs were previously explained by immunologic mechanisms that nonetheless await direct demonstration. Worsening of NYHA III-IV class HF by infliximab was tentatively attributed to complement fixation on cardiomyocytes that expressed TNFα-TNFR complexes, similar to what occurs in post-ischemic heart that expresses self-antigens and evokes autoimmune myocarditis (43). In the case of etanercept, however, a more complex and self-repeating mechanism has been tentatively put forward: rapid binding of TNFα to etanercept and equally rapid or faster dissociation of TNFα results in longer-lived circulating levels TNFα in a biologically active form (43). Both mechanisms may be valid if one only looked at the high levels of TNFα in the bloodstream and considered anti-TNFα drugs from immunologic viewpoints only.
We suggest that the failing heart is a pharmacologic sanctuary where ischemia or hemodynamic overload triggers an autocrine loop that protects cardiomyocytes from attack by circulating high TNFα. The loop begins with cardiomyocyte synthesis and release of discrete amounts of TNFα, and proceeds with the binding of such TNFα to TNFR1-TNFR2 and activation of downstream survival-oriented cell responses. Potentially toxic “high” TNFα that came from systemic dysregulation of cytokine sources (44) and penetrated the sanctuary would find cardiomyocytes protected by preconditioning (Figure 2A); in contrast, treatment with infliximab or etanercept disrupts the autocrine loop and leaves cardiomyocytes unprotected against high TNFα (Figure 2B). In progressive HF, altered permeability of coronary microvasculature should, in fact, be accompanied by an increasing penetration of the sanctuary by high molecular mass anti-TNFα drugs (45, 46).
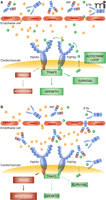
A. Cardiomyocytes may synthetize and release discrete amounts of TNFα that bind to TNFR2 and eventually pass to TNFR1, activating survival-oriented cell responses that protect against higher levels of TNFα. B. Penetration of anti-TNFα drugs through discontinuous endothelium of the failing heart interrupts the autocrine loop and eventually shifts TNFR1–2 to signaling apoptosis. Solid blue circles represent circulating TNFα; solid green circles represent TNFα released by cardiomyocytes. INF, infliximab; ETA, etanercept; TNFR1–2, TNFα receptor 1–2; TRAF2, TNFα receptor–associated factor 2; TRADD, TNFR1-associated death domain.
The story we suggest is in part reminiscent of cardiotoxicity from trastuzumab, a humanized epidermal growth factor receptor 2 (ErbB2)-specific antibody that was developed to kill ErbB2-overexpressing breast cancer cells by antibody-dependent cell-mediated cytotoxicity (ADCC) (46). Trastuzumab causes HF by binding to ErbB2 that is constitutively expressed in cardiomyocytes, but cardiotoxicity is not mediated by ADCC: trastuzumab halts survival signals that are relayed by ErbB2 and helps cardiomyocytes to withstand toxicity from other drugs or hemodynamic overload or other comorbidities (7, 47, 48). In principle, there are other intersections or similarities between our story and the lessons from trastuzmab. ErbB2 activation makes the heart more responsive to vagal stimulation (49), which is known to increase TNFR2 expression and TNFR2-mediated protection against ischemia-reperfusion (25). Furthermore, patients with HF exhibit circulating levels of soluble forms of both TNFR1–2 and ErbB2 (50); unfortunately, however, uncertainties about the mechanisms of receptor shedding and the pathophysiologic role of circulating receptors do not justify much speculation in this setting. With that said, the time is right to explore anti-TNFα drugs from the broader perspective of other drugs that decrease cardiotrophic signals.
Anti-TNFα drugs have now been used in more than two million patients with RA or other chronic inflammatory diseases. Investigational indications ranging from Kawasaki’s disease to steroid-refractory acute graft-versus-host disease and dermatomyositis (or polymyositis) or many others will conceivably surface in clinical practice in the next years and will further expand the marketing of anti-TNFα drugs. Concerns about cardiac events should guide or limit, when appropriate, the prescribing of these agents.
In this article we have challenged facts and controversies about cardiotoxicity induced by anti-TNFα drugs. Looking at TNFα as a Janus-like molecule that brings life or death to cardiomyocytes as a function of its levels in cardiac sanctuaries or in the bloodstream may help drug developers, pharmacologists, and clinicians to redefine the appropriateness and safety of these drugs in a given patient or another. Defining the cardiovascular risk of an individual should not be confined to measuring plasma levels of TNFα but probably should be extended to probe the capability of the heart to capture trophic signals from “some TNFα” and other factors, whether in absence or presence of anti-TNFα drugs. These concepts call for diagnostic measures or provocative tests that can only be surmised at this time. We conclude that anti-TNFα drugs should be included in the list of potentially cardiotoxic drugs. Putting these drugs on that list calls for research—and good sense—that go from bench to bedside.
Acknowledgments
Work in the authors’ laboratories was supported by University Campus Bio-Medico Special Project “Cardio-Oncology” (to GM).
- Copyright © 2011
References
Fabio Cacciapaglia, MD, received his degree in medicine (2004) and boarded in clinical immunology (2008) at the University Campus Bio-Medico of Rome. He currently attends a Research Doctorate Program in Sciences of Organ Plasticity and Tissue Regeneration for Functional Recovery at the University Campus Bio-Medico of Rome. A member of the Italian Society of Rheumatology, he is an attending physician of Internal Medicine at the “N. Melli” Hospital of San Pietro Vernotico (Brindisi) and participates in pre-clinical and clinical trials of anti-rheumatic therapies with TNFα targeted drugs or disease modifying anti-rheumatic drugs. E-mail f.cacciapaglia{at}unicampus.it; fax +39-0831-670377.

Pierantonio Menna, PhD, graduated in pharmaceutical chemistry in 2000 and was granted a doctoral research degree in 2005 from the G. d’Annunzio University of Chieti School of Pharmacy and the Center of Excellence on Aging. From 2004 to 2005, he was Visiting Scientist at the Cancer Pharmacology Center of the University of Pennsylvania School of Medicine, Philadelphia. He is presently a Clinical Pharmacologist at the University Hospital of Campus Bio-Medico of Rome. His main interest is in drug metabolism, therapeutic drug monitoring, and investigational pharmacokinetics and biomarkers.

Luca Navarini, MD, works in the Department of Clinical Medicine and Rheumatology at the Campus Bio-Medico University of Rome. His research is focused on cardioprotection and cardiotoxicity induced by TNFα, in collaboration with the laboratory of Drug Sciences of the same Institute.
Antonella Afeltra, MD, is Professor of Rheumatology and Chief of Clinical Medicine and Rheumatology at the University Campus Bio-Medico of Rome. Her scientific areas of interest include the pathogenesis of systemic autoimmune diseases and their cardiovascular involvement, the pathophysiology of neuropsychiatric disorders in connective tissue diseases, and the role of biologic agents in the treatment of arthritis.

Emanuela Salvatorelli, PhD, graduated in pharmacy in 2000 and was granted a doctoral research degree in 2005 from the G. d’Annunzio University of Chieti School of Pharmacy and the Center of Excellence on Aging. She is presently Assistant Professor of Pharmacology and Head of Laboratory at the Center for Integrated Research of the University Campus Bio-Medico of Rome. Her main interest is in drug metabolism and bioactivation mechanisms of toxicity. She developed translational models of human heart to probe the metabolic determinants of cardiotoxicity from antitumor anthracyclines and investigational analogues.

Giorgio Minotti, MD, PhD, is Professor of Pharmacology and Chief of Clinical Pharmacology at the University Campus Bio-Medico of Rome. He graduated in medicine in 1981 and boarded in oncology in 1984. He was a Fulbright Scholar at the Department of Biochemistry of Michigan State University, Lansing (1985–1987) and a Fulbright Visiting Professor at the Department of Physiology and Biophysics of Case Western Reserve University, Cleveland. He is member of the Italian Society of Pharmacology, the Italian Society of Toxicology, the American Society of Oncology, and the American Society of Pharmacology and Experimental Therapeutics, and an honorary member of the International Society of Cardio-Oncology. His main research interest is in preclinical models and clinical correlates of cardiotoxicity from antitumor drugs and other noncardiovascular drugs.


