- Institution: Stanford Univ Med Ctr Lane Med Lib/Periodical Dept/Rm L109
- Sign In as Member / Individual
Vasomotion: Mechanisms and Physiological Importance
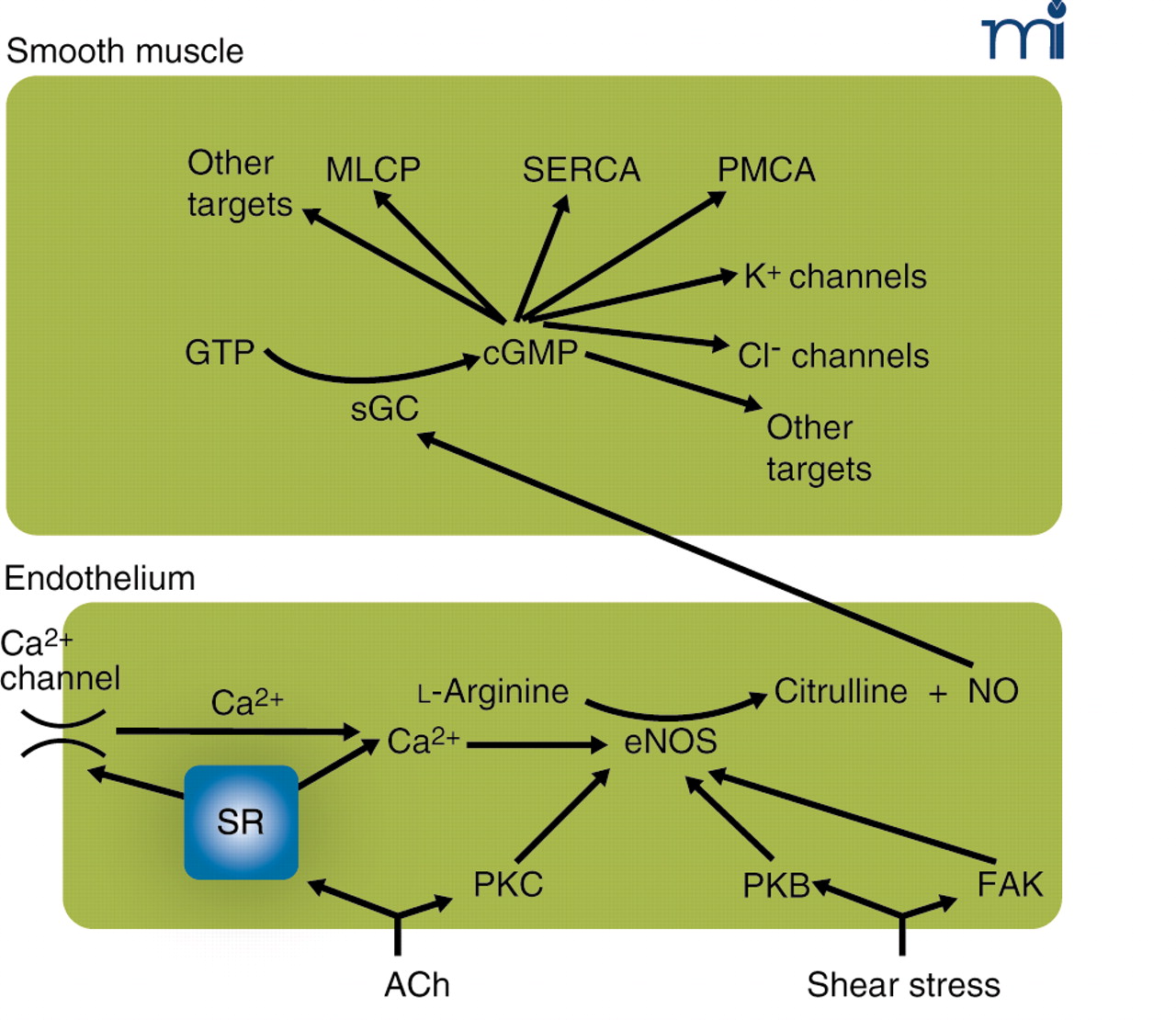
The endothelium–NO–cGMP axis. Stimulation of the endothelium, either by receptor agonists such as acetylcholine (ACh) or by mechanical forces, activates endothelial nitric oxide synthase (eNOS). This may occur by calcium-dependent and calcium-independent pathways, some of which are indicated here. Calcium elevation may be caused by release from stores or by influx through calcium-permeable pathways [e.g., transient receptor potential (TRP) channels] activated as a consequence of store depletion; the mechanism by which store depletion activates influx is subject to intense study. Calcium-independent pathways involve the activation of various kinases. Activated eNOS catalyzes the conversion of l-arginine to citrulline and nitric oxide (NO), which diffuses to the smooth muscle where it activates soluble guanylate cyclase (sGC), converting GTP to cyclic GMP (cGMP). In the smooth muscle, cGMP has a multitude of effects that comprise activation of myosin light-chain phosphatase (MLCP) causing desensitization to intracellular calcium of the contractile machinery, probably activating the sarcoplasmic reticulum calcium ATPase (SERCA) and the plasma membrane calcium ATPase (PMCA), opening of K+ channels, and, as suggested by our recent experiments, enabling a calcium-activated Cl− channel. Many of these effects are mediated by activation of protein kinase G (PKG). Most effects of NO on the smooth muscle inhibit contraction; the Cl− channel, which causes depolarization and thus promotes contraction, we suggest is responsible for superimposed intermittent contractions responsible for vasomotion. PKB, protein kinase B (Akt); PKC, protein kinase C; FAK, focal adhesion kinase.

