Vasomotion: Mechanisms and Physiological Importance
Abstract
Vasomotion, rhythmic oscillations in vascular tone caused by local changes in smooth muscle constriction and dilation, has been observed since the invention of the microscope, but its physiological role and its underlying mechanism remain elusive. Present in many tissues, vasomotion might serve as a protective mechanism under conditions of ischemia. Its generation seems dependent on synchronization of a multitude of individual cellular oscillators. The concept of calcium release and uptake by the smooth muscle calcium stores, synchronized by sarcolemmal ion channels, and sometimes influenced by endothelial factors, may provide further understanding of this little-understood phenomenon.
Introduction
Vasomotion is an oscillation of vascular tone that is generated from within the vascular wall and is not a consequence of heartbeat, respiration, or neuronal input. Vasomotion is a phenomenon observed by many scientists studying vascular physiology, occurring both in vivo and in vitro (Figure 1⇓) in many vascular networks if the correct conditions are present. The first detailed report of vasomotion—in the bat wing—was made more than 150 years ago (1) .
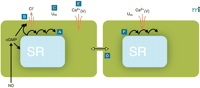
Conceptual model for the generation of vasomotion in rat mesenteric small arteries. The basic oscillator is the sarcoplasmic reticulum (SR), which intermittently releases calcium leading to regenerative waves of calcium traversing the cell (A). In the presence of cyclic GMP (cGMP), this calcium also is able to activate a chloride channel in the plasma membrane (B). If this current is activated in a sufficient number of cells at the same time, cells will depolarize (C). Also cells that are not contracting at that moment will depolarize, because all cells are electrically coupled (D). Depolarization will cause calcium influx through L-type calcium channels (E), which not only triggers contraction but also calcium release in quiescent cells (F), thus resetting their intracellular oscillators. In this way, the likelihood for a sufficient number of cells becoming active together in the next cycle is very high, and the cells in effect become phase-locked. cGMP promotes vasomotion by its permissive action on the ion channel, most likely via protein kinase G–mediated phosphorylation of the channel, but inhibits vasomotion by actions on the SR by influencing intracellular calcium availability, uptake, and/or release. Differences in this balance may explain the variable influence of endothelium in vasomotion. Ca2+(V), voltage-gated (L-type) calcium channel; Um, membrane potential.
In spite of this relatively long history, two important questions remain incompletely understood: What is the cellular mechanism responsible for vasomotion? What is the physiological consequence of vasomotion?
In many experimental contexts vasomotion is problematic; for example, it is difficult to define specific amounts of tone in an oscillating vessel and therefore to determine concentration-response curves unambiguously. Furthermore, vasomotion is frequently unpredictable and difficult to reproduce; occasionally, experimental animals cease to exhibit vasomotion for varying periods of time, and in vitro experiments fail because vasomotion cannot be replicated. Nevertheless, during the last twenty years, much progress has been made with respect to the questions of cell-specific mechanisms and physiological consequences of vasomotion. This review briefly summarizes the hypothesized mechanisms of vasomotion and discusses some of the physiological and pathophysiological roles suggested for vasomotion.
Occurrence of Vasomotion
Vasomotion has been described both in the intact animal and in isolated arteries. A summary of the literature on intact animals was made by Funk and Itaglietta (2) beginning with the observations made by Jones in 1852 of rhythmic changes in the bat wing (1) through to the early 1980s. Apart from direct demonstrations of diameter changes in vessels, the presence of vasomotion has also been demonstrated by measuring blood-cell velocity (3) , capillary pressure (flow motion) variations (4) , or laser-Doppler flow signals (Box 1) (5) . The presence of localized oscillations in blood flow in human cutaneous circulation has been taken as evidence of the local, non-nervous origin of the oscillations. In hamsters, vasomotion has been carefully studied in skin-fold preparations from both awake and anesthetized animals (6) . Vasomotion was observed in the awake animal but disappeared readily upon anesthesia with pentobarbital and chloralose–urethane (6) . Similarly, in the cerebral circulation of rabbits, vasomotion is observed under wakeful conditions and is depressed by anesthesia (7).
Methods used in the study of vasomotion.
-
Vital microscopy . Direct observation of the movements of blood vessels has been the traditional method to study vasomotion. While mostly done in anesthetized animals, implanting a skin window (6) has allowed investigators to inspect blood vessels in underlying tissue in the awake animal.
-
Pressure measurements. Measuring the pressure in the capillaries after direct cannulation gives a measure of the ratio of pre- to post-capillary resistance, provided the arteriovenous pressure difference is constant throughout the measurement. The most likely cause of fluctuations in this ratio is changes in the degree of constriction of precapillary resistance vessels.
-
Blood-cell velocity measurements. The time it takes a cell to pass a certain distance along a capillary is proportional to the volume flow through the capillary as long as capillary diameter stays constant (a reasonable assumption under most conditions). With the same provisos as for pressure measurements, variations in flow reflect upstream constrictions, since flow is determined by the pressure gradient along the capillary.
-
Laser-Doppler flow measurement. Light from a strong, monochromatic light source undergoes a frequency shift (Doppler shift) when it is backscattered from moving objects; analysis of this shift in backscattered light from living tissue gives a relative measure of blood cell movement, thus average blood flow in a volume of tissue. This volume is typically hard to define; in other respects the technique is similar in principle to the blood-cell velocity measurement.
-
Isolated vessels . Studies of excised artery specimens with various kinds of organ-bath techniques have the advantage of examining strictly local phenomena under well-controlled conditions. For technical reasons these studies are limited to relatively large arteries, not those that typically are responsible for capillary flow motion in vivo . Still, they may provide some insight into the cellular mechanisms that can generate oscillations.
In skeletal muscle, vasomotion can be induced after bleeding the animal (8) , but recent reports have indicated that vasomotion is not present under more physiological conditions (8–10). Thus, if anesthesia is very well-controlled in terms of blood pH and blood gases, vasomotion is not observed (in the tenuissimus muscle), whereas vasomotion is observed in one-third of animals whose anesthesia was administered without this precise control (9) . Metabolic acidosis by the infusion of dilute HCl solution induces vasomotion in most animals. In support of the idea that vasomotion is not present under physiological conditions, implanted flow probes in the triceps muscle of awake rabbits under control conditions failed to report any vasomotion (11) . One must thus conclude that in this vascular bed vasomotion is initiated by metabolic stress and is absent under resting conditions. This is in contrast to the early reports on human skin and hamster subcutaneous muscle, where vasomotion was reported under conditions that can hardly be denoted as pathological. The problem with these latter reports is that the prevalence of vasomotion was not systematically reported, suggesting that vasomotion might have been present only for brief periods in some of these studies. Schmidt has critically reviewed the question of whether vasomotion is present under physiological conditions (11) and reports that vasomotion is generally most prevalent under conditions of reduced perfusion. Thus, we may conclude that possibly both the presence as well as the intensity or quantity of vasomotion is highly variable. The question of whether vasomotion occurs under normal conditions or only in circulatory stress is at present being examined.
Oscillations in the diameter or tone of large isolated arteries have also been observed in vitro with a wide variety of oscillatory patterns, depending on the invitro technique. Whether such oscillations should be denoted vasomotion, suggesting a similar origin as that seen in microcirculation, is a matter of debate. However, human coronary (12) and pial (13) arteries, canine subcutaneous arteries (14) , rabbit ear small arteries (15) , rabbit mesenteric artery (16) , rat basilar artery (17) , carotid artery from rat or dog (18, 19) , rat tail artery (20) , rat mesenteric small arteries (21) , rat irideal arterioles (22) , and many other preparations display some kind of oscillation in vitro, either spontaneously or when activated by an agonist (e.g., noradrenaline). These preparations have been used as models to understand the cellular mechanisms underlying vasomotion.
Neurohumoral Influence on Vasomotion
In situ oscillations in the vasculature arise from many different causes. Mechanical oscillations from the movements of the heart and respiration are most pronounced in veins, but respiratory movements may confound vasomotion measurements by the laser-Doppler technique (23) . Oscillations in nervous discharges—with rhythms such as Traube-Hering-Mayer waves (Box 2) having a periodicity of ten seconds—directly generate oscillations in vascular tone that could be mistaken for vasomotion. However, because these oscillations arise from variations in neural discharge, they would be synchronous in large parts of the body in contrast to true vasomotion, which is a local phenomenon. Thus, if only a local area is under study, it becomes difficult to distinguish systemic vascular oscillations from local oscillations. During measurements of cutaneous vasomotion in vivo, changes in tone inevitably contribute to the observed flow changes (24) and possibly are predominant in larger arteries (25) . However, the presence of localized changes in flow (26) indicates that not all changes in tone are caused by external signals to the vessels but are generated locally.
Blood pressure oscillations: Traube-Hering-Mayer waves.
In the 19th century, interest began to focus on various oscillations observed in arterial blood pressure. One began to talk about oscillations of different order. Order 1, the fastest, were oscillations due to heartbeat. Order 2 were oscillations due to the mechanical movements of the chest during respiration. Order 3 referred mostly to oscillations slower than the other two and were of unknown origin. However, there were also some waves of the same frequency as order 2 that could not be explained as mechanical consequences of breathing.
Traube (98) described in 1865 that if artificial ventilation in a curarized animal was stopped, blood pressure rose and started to oscillate at a frequency of about seven rhythms/min (similar to 2nd order oscillations). Similarly Hering (99) in 1869 could show that if, also in the curarized animal, ventilation was made rapid and shallow, similar blood pressure waves emerged. In 1876, Mayer (100) reported that in spontaneously breathing animals, blood pressure oscillations with a frequency lower than that of respiration appeared if respiration was slightly hindered (therefore 3rd order waves by definition but actually rather similar to the Traube and Hering waves). It now appears that all these oscillations can be ascribed to oscillations in efferent vasoconstrictor nerve activity, which can be evoked or exaggerated by cerebral hypercapnia (abnormally high concentrations of carbon dioxide in the circulating blood).
Observations on isolated arteries indicate that vasomotion is enhanced in amplitude and/or frequency by vasoconstricting receptor agonists. For instance, rat mesenteric small arteries do not exhibit vasomotion under basal conditions because of a lack of tone, but if noradrenaline is administered exogenously, contraction occurs and oscillations of the contractions develop on top of the induced tone (21) . Such an effect is not limited to noradrenaline, but is observable in the same assay in the presence of vasopressin or neuropeptide Y ( Gustafsson and Nilsson, unpublished results; see also 27) , as well as other vasoactive peptides (28–30) , suggesting it is primarily a matter of activating the muscle rather than a specific effect from a certain receptor.
The observations on isolated arteries positively correlate with observations on the microcirculation in hamster skin-fold preparations, where adrenergic inhibition reduces, and adrenergic stimulation enhances, vasomotion (31) . The enhancing effects of systemic hypoxia on vasomotion in this preparation were prevented by phentolamine (32) , indicating that sympathetic tone is a prerequisite for vasomotion, either by providing a basal level of tone necessary for oscillation or by stimulating the oscillations directly.
The fact that noradrenaline and other agonists may stimulate vasomotion might explain the induction of vasomotion by hemorrhage (8) , in that the ensuing sympathetic activation may provoke vasomotion. Consistently, it has been reported that inhibiting sympathetic input to the muscle eliminates previously observed slow-wave vasomotions (11, 33) . Furthermore, after severing the sciatic nerve in rabbits, vasomotion could be induced in the tenuissimus muscle by stimulation of the nerve, although only when femoral artery pressure was reduced (11) . Infusion of adrenaline, noradrenaline, or vasopressin in this preparation was similarly capable of eliciting vasomotion at reduced blood pressure (11) . A somewhat different observation comes from Rücker et al., who studied vasomotion using controlled perfusion of pedicled osteomyocutaneous flaps (superficial tissue flaps consisting of skin, muscle, and bone excised from the animal in such a way that they are connected only by a narrow stalk through which they obtained their nerve and blood supply) (34) . Although no tissue exhibited vasomotion under basal conditions, flow reduction did induce vasomotion in the muscle, irrespective of whether innervation was intact or not. Thus, the inducing stimulus of vasomotion might be flow reduction, either in itself or via metabolic changes in the tissue. However, the sympathetic nervous system might still facilitate vasomotion, because the neural activity under these experimental conditions would probably have been low, and removing its influence would therefore not change the situation significantly.
That nerves are not necessary for oscillations in vascular tone has also been supported by many studies on isolated arteries. These are studies where either the generation of action potentials in the vasomotor nerves (17, 35) or the release or action of the transmitters (22, 36 , 37) have been blocked.
Taken together, these studies seem to indicate that neural influence is not required for vasomotion to occur. Vasomotion requires some degree of tone, and if spontaneous tone is not sufficient, neural or humoral input may elevate tone to a level where vasomotion may occur. It is also possible that these inputs may modulate the oscillating mechanism in the vessels, thereby influencing vasomotion frequency or phase.
In this context it is interesting that vasomotion can be induced in situations of reactive hyperemia—the increase above baseline in blood flow after temporary flow obstruction (26) —demonstrating that tone in arterioles is not fully eliminated by the accumulation of metabolites during the ischemic period. Furthermore, the upstream vasodilation that mediates the hyperemia would cause an elevation of precapillary pressures. Because vasomotion can be triggered by reduced perfusion, which would cause pressure reduction, it does not appear likely that it is the acute pressure change itself that is the stimulus for vasomotion. Conceivably, the accumulation of metabolites, with ensuing pH changes etc., during the ischemic period in this situation as well as during hypoperfusion is the critical event.
On the Mechanism of Vasomotion
Vasomotion requires coordination of the activity of the vascular smooth muscle cells in the vessel wall. There is little doubt that this is achieved through electrical communication—electrical communication may be the only means of communication fast enough to coordinate responses over several millimeters of vessel—and that oscillations in membrane potential are responsible for the oscillations in vessel tone. That is, whenever membrane potential has been measured during vasomotion, the membrane potential either oscillates with the same frequency as the vasomotion (19, 21, 22, 36) or shows bursts of action potentials in phase with the tension oscillations (13, 38) , and the changes in membrane potential seem to precede the oscillation in vessel tone (21, 22). One exception is the observation by Haddock et al. (39), who show that an oscillating current can be observed in cells in pinned-out short segments of irideal arterioles under voltage clamp, and under these conditions “[v]isual observation of the preparation indicated that rhythmical movements of the preparation continued whilst the voltage clamp was applied.” It is, however, not clear how such movements were propagated, and in a further study the authors suggest a model based on electrical coupling of cells (40).
Further support for the importance of membrane potential changes in the coupling of cells in vasomotion comes from studies using gap-junction blockers, which would inhibit vasomotion if electrical coupling through gap junction was synchronizing cellular oscillations. This was actually observed by Chaytor et al. (41) who developed peptides mimicking connexin loops to break connections between gap-junctional hemichannels. These authors used the disappearance of vasomotion as an argument for the peptides’ activity on gap junction; it still remains to be shown whether these peptides inhibit electrical coupling between cells and that their effect on vasomotion is not unspecific. It is interesting to note that another gap-junction inhibitor, 18α -glycyrrhetinic acid, inhibited both mechanical and electrical oscillations in irideal arterioles (22) —the effect on electrical oscillations in single cells suggests either a more profound role for membrane potential changes in the generation of oscillations or an unspecific effect of this compound. Another putative gap junction inhibitor, heptanol, has been shown to have important non-specific effects (42, 43).
It has been suggested that the electrical activity that governs vasomotion is paced by specific pacemaker cells situated near bifurcations of blood vessels (44, 45) . This concept has been supported by the demonstration of increased L-type channel density on the surface of vascular smooth muscle cells and an apparent spread of the increased [Ca2+ ]i , as measured by fluorescent imaging, from branch points to other parts of the vessel following KCl depolarization (44) . On the other hand, there have been very few reports (46–48) on smooth-muscle-like cells having special morphological or biophysical characteristics that in the vascular wall could mediate a pacemaker-like function similar to that of Cajal cells in the intestine (49) . However, Cajal cells were very recently identified in the portal vein (50) , allowing for the intriguing possibility that some cells in the vessel wall are more likely to “drive” vasomotion. For such a scenario to be relevant, vasomotion should originate from the same site in the vasculature or the same position in an isolated vessel whenever vasomotion is initiated. Due to the rapid spread (presumably > 1 cm/sec) of the electrical signal, it is experimentally difficult to determine whether this occurs, and to our knowledge, it has not been demonstrated. Other studies have indicated that vasomotion is unlikely to start from a fixed pacemaker (51, 52).
An alternative possibility is that coordination occurs through entrainment of smooth muscle cells oscillating asynchronously. In other words, it is possible that all or nearly all vascular smooth-muscle cells may be able to oscillate and, through interconnections, eventually entrain. With this scheme it will be of interest to: a) understand the mechanism responsible for the oscillation in the individual cell and b) understand how the coupling of the cells leads to entrainment.
An intriguing possibility for an oscillating mechanism was suggested by Siegel (19) and is based on oscillations in phosphofructokinase-1 activity leading to oscillations in ATP and, consequently, oscillations in Na+ , K+ –ATPase (or Na+ pump) activity. The electrogenic nature of the Na+ pump would lead to oscillations in membrane potential, which could spread to neighboring cells. This suggestion is supported by the observation of several authors that ouabain-mediated inhibition of the Na+ pump blocks vasomotion (35, 53–56). How the Na+ pumps of individual cells are entrained in this model to effect synchronized vasomotion in larger areas has, however, not been discussed.
Several authors have suggested that the sarcoplasmic reticulum (SR) is important for vasomotion (22, 56–59). Different blockers of the SR Ca2+ -pump (SERCA) inhibit vasomotion, and the interaction of an oscillator whose activity is mediated by the SR and the sarcolemma of smooth muscle cells has been suggested (51, 58, 60). We have hypothesized (36) that the unsynchronized waves of Ca2+ reported in several smooth muscle cells—both isolated cells (61, 62) and in the intact arterial wall (36, 62–64) —might constitute a basal oscillator, which, through cooperation with the sarcolemma, leads to membrane potential oscillation and consequent synchronization of smooth muscle cells in the vascular wall (Figure 1⇑). Recently, Haddock and Hill (40) proposed a model based on similar principles (calcium release from intracellular stores synchronized by a calcium-dependent chloride current) to account for vasomotion in irideal arterioles.
Another important element in the mechanism responsible for vasomotion is the endothelium–nitric oxide (NO)–cGMP axis (Figure 2⇓). Although little evidence has been obtained from in vivo investigations (65, 66) , several authors have assessed the role of these elements for vasomotion under in vitro conditions. Strikingly, the reports differ quite substantially, and although NO prevents vasomotion in some arteries (22, 67) , NO enhances the prevalence of vasomotion in other vessels (21, 37) . An understanding of the reason for this variable effect may bring us closer to an understanding of the mechanisms involved in vasomotion.
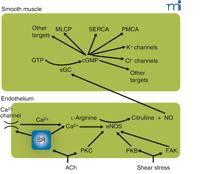
The endothelium–NO–cGMP axis. Stimulation of the endothelium, either by receptor agonists such as acetylcholine (ACh) or by mechanical forces, activates endothelial nitric oxide synthase (eNOS). This may occur by calcium-dependent and calcium-independent pathways, some of which are indicated here. Calcium elevation may be caused by release from stores or by influx through calcium-permeable pathways [e.g., transient receptor potential (TRP) channels] activated as a consequence of store depletion; the mechanism by which store depletion activates influx is subject to intense study. Calcium-independent pathways involve the activation of various kinases. Activated eNOS catalyzes the conversion of l-arginine to citrulline and nitric oxide (NO), which diffuses to the smooth muscle where it activates soluble guanylate cyclase (sGC), converting GTP to cyclic GMP (cGMP). In the smooth muscle, cGMP has a multitude of effects that comprise activation of myosin light-chain phosphatase (MLCP) causing desensitization to intracellular calcium of the contractile machinery, probably activating the sarcoplasmic reticulum calcium ATPase (SERCA) and the plasma membrane calcium ATPase (PMCA), opening of K+ channels, and, as suggested by our recent experiments, enabling a calcium-activated Cl− channel. Many of these effects are mediated by activation of protein kinase G (PKG). Most effects of NO on the smooth muscle inhibit contraction; the Cl− channel, which causes depolarization and thus promotes contraction, we suggest is responsible for superimposed intermittent contractions responsible for vasomotion. PKB, protein kinase B (Akt); PKC, protein kinase C; FAK, focal adhesion kinase.
In regard to the role of NO in enhancing vasomotion, we have recenty discovered a cGMP-dependent, Ca2+ -activated chloride channel (Matchkov, Aalkjær, and Nilsson, unpublished results). This channel appears responsible for coupling the Ca2+ oscillations generated by the SR to the membrane current that synchronizes individual cells. We hypothesize that NO levels could elevate cGMP concentrations in smooth muscle cells, thereby increasing the likelihood of synchronized oscillation. At the same time, other effects of NO may be inhibitory on the SR oscillator (68) by modulating intracellular calcium availability, uptake, and/or release. Thus, an NO–cGMP system might regulate vasomotion both through the generation of oscillation and cellular coupling.
Functional Consequences of Vasomotion
There is very little data concerning the physiological function of vasomotion. It is conceivable that vasomotion is merely a harmless consequence of smooth-muscle contraction, oscillating under particular conditions, that has not been eliminated during evolution. Flow oscillation might arise from oscillations based on the phase delay in the myogenic response (69) ; if the response of a blood vessel to a pressure change comes with a delay, a brief pressure pulse may start an oscillation in vessel tone. On the other hand, vasomotion may be beneficial under certain conditions. A vessel with an oscillating diameter has higher conductance (and therefore greater flow) than a vessel with a constant diameter of the same average width (70) . An oscillating contraction might be less costly in terms of energy usage than a constant contraction, but experimental data supporting such a view are scarce. Along similar lines, one could argue that by constantly varying its state of contraction, the vascular muscle does not enter the “latch state” of contraction. (The latch state, characteristic of many smooth muscles, is a state of low contraction velocity combined with low ATPase activity, enabling the muscle to maintain contraction with low energy expenditure at the expense of the ability to shorten rapidly). On the other hand, avoidance of the latch state might confer the advantage that the muscle remains able to shorten rapidly, thereby capable of responding quickly to neurogenic commands.
One of the most interesting suggestions is that vasomotion is beneficial for tissue oxygenation, especially in situations where perfusion is critically limited. The foundation for this idea comes from theoretical modeling by Tsai and Intaglietta (71) that suggests that periods of high flow at low frequency of oscillation, such as during the slow-wave vasomotion typical for proximal arterioles, permit long-distance diffusion of oxygen to capillaries; thus, at critical perfusion levels, oxygen becomes more homogeneously distributed, and less of the tissue is exposed to very low levels of oxygen. However, the recent theoretical analysis by Goldman and Popel (72) argued that vasomotion could promote tissue oxygenation only at low concentrations of myoglobin, suggesting that vasomotion would have limited influence on muscle oxygenation. To address the relation of vasomotion to tissue oxygenation, Rücker et al. (73) studied perfused rat hindlimbs by intravital microscopy and used NADH fluorescence as an indication of the metabolic state of the tissues. When perfusion was reduced, vasomotion was induced in most muscle preparations. By comparing preparations with and without vasomotion, the authors concluded that vasomotion enhanced blood flow to adjacent tissues (e.g., skin, subcutis, and periosteum) and maintained high NADH fluorescence in the tissues adjacent to muscle, suggesting a beneficial effect on oxygenation in adjacent tissues. Thus, vasomotion may be of little importance to muscle (with a high cellular buffering capacity for oxygen) but of greater importance for other tissues. This result seemingly suggests that flow is diverted from the muscle to the other tissues, which is not consistent with that fact that vasomotion by itself reduces flow resistance; but the results could imply that when vasomotion is present, artery tone is higher in the muscle than when it was absent.
Some theoretical models (71, 74) have been constructed regarding the role of vasomotion in capillary function, particularly in the maintenance of both low interstitial pressure and contancy of paracapillary fluid flux. These models have so far not given unanimous predictions, but it seems clear that vasomotion may strongly influence capillary exchange. Unfortunately, the conclusions borne from theoretical studies have received little direct support from experiments (71, 74).
Pathophysiological Role of Vasomotion
There is evidence that the prevalence of vasomotion may be altered under pathophysiological conditions. In diabetes there is good evidence that vasomotion is less prevalent. Stansberry et al. (75) found that in insulin-dependent and in insulin-independent diabetics the amplitude of vasomotion in the finger was reduced to about 20%, and the authors concluded that vasomotion was impaired in 75% of diabetics. In the skin of the feet of diabetic patients with neuropathy, but not in diabetics without neuropathy, the amplitude of flow-motion is also reduced (76) . These observations are in accordance with a number of animal studies showing that in streptozotocin-induced diabetes, vasomotion essentially disappears in bat wings (77) , hamster cheek pouch (78) , and rat spinotrapezius muscle (79) in vivo. Renaudin et al. reported that acute infusion of glucose to rats increased the prevalence of vasomotion in the spinotrapezius muscle (79). We are unaware whether any research has addressed the pathophysiology of vasomotion under in vitro conditions.
Metformin, the biguanide that is used to treat non-insulin-dependent diabetes, increases the prevalence of vasomotion. Intravenous injection of metformin into hamster cheek pouches acutely induces vasomotion (80) , and vasomotion reappears in the wings of diabetic bats treated with metformin (77) . Also, in anesthetized hamsters exposed to hemorrhagic shock, injection of metformin acutely induced vasomotion (81). Similarly, in mildly hyperglycemic animals (streptozotocin treated), injections of metformin prevented insulin-induced inhibition of vasomotion (78) . As for the studies on diabetes, there are no reports on the effect of metformin in isolated arteries, and the reason for this effect of metformin is entirely unknown.
Increased prevalence of vasomotion has been extensively studied in hypertension. Enhanced vasomotion has been observed in femoral arteries from renal hypertensive rats (82), deoxycorticosterone acetate– (DOCA)–salt hypertensive rats (83) , and in rats genetically predisposed to hypertension (35, 38, 83–87). Interestingly, studies in cross-bred genetic hypertensive and normotensive rats (88, 89) show that the prevalence of increased vasomotion positively correlates with increased blood pressure, indicating a tight coupling between high blood pressure and the ability to oscillate. These findings, together with the observation that vasomotion seems to be less prevalent in hypertensive rats treated with ACE inhibitors (90, 91), seem to indicate that high blood pressure evoked through several different mechanisms may induce the oscillations. Thus, in contrast to acute pressure changes, a chronically elevated pressure level apparently promotes vasomotion. One could thus speculate that the capillary rarefaction seen in the hypertensive vasculature (92) could be ameliorated by vasomotive oxygen delivery (see above). Although an in vivo study of renal hypertensive rats reveals periods of relative vasoconstriction and vasodilation in the cremaster muscle (93) , this pattern of contraction and dilation is not similar to what is normally reported for vasomotion. Nevertheless, in essential hypertensive patients, Hollenberg and coworkers (94, 95) observed increased amplitude in vasomotion per se. There is also some in vitro evidence for enhanced vasomotion in human hypertension: isolated arteries from women with preeclampsia exhibit more vasomotion than is observed in arteries from normotensive pregnant women (96, 97).
Conclusion
Vasomotion, in the restricted sense of the word, denotes oscillations in vascular tone. These oscillations can occur in vessels in many tissues in vivo, and correlates can be seen in vitro in isolated preparations of arteries. The in vitro approach enables us to study the mechanisms underlying oscillations in multicellular tissues, but the connection between these generally relatively large preparations and the oscillations in terminal arterioles seen in vivo remains to be determined. In spite of a large body of experimental work, the physiological significance of vasomotion remains elusive. Arguably, there is some evidence pointing in the direction of a role in tissue oxygenation when perfusion is compromised. If so, the phenomenon would be a mechanism of substantial pathophysiological importance. Although vasomotion has been like the weather—many talk about it but few do anything seriously about it—we have begun to unravel the cellular and molecular mechanisms of oscillation and cellular coupling underlying vasomotion.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Holger Nilsson, MD, DMSc, is an associate professor and Christian Aalkjær, MD, DMSc , is professor at the Department of Physiology, University of Aarhus. Both are members of the Water and Salt Research Center at the university. Address correspondence to either HN or CA. E-mail holger.nilsson{at}fi.au.dk; ca{at}fi.au.dk; fax +45 86129065.

