LQT4 Gene: The “Missing” Ankyrin
Significant advances from molecular genetic studies in the past decade have provided remarkable insights into the pathogenic mechanisms of long QT syndrome (LQTS). The syndrome is characterized by prolongation of the QT interval (Figure 1⇓) and T wave abnormalities as observed on electrocardiograms (ECGs). Patients with LQTS are susceptible to ventricular tachyarrhythmias (VT), or torsade des pointes (TdP), that may degenerate into ventricular fibrillation (VF) and lead to syncopal episodes and sudden death (1). There are two inherited forms of LQTS: autosomal dominant LQTS with normal hearing, and the much rarer autosomal-recessive LQTS associated with congenital deafness (1). The incidence of LQTS for both inherited forms has been estimated to be about 1 per 10,000 without an apparent ethnic or geographic predilection (2, 3).
In 1995, two back-to-back studies published in Cell reported on the LQT2 gene [referred to as HERG (human ether-a-go-go, KCNH2)] on chromosome 7q35–36 (4) and the LQT3 gene (SCN5A) on 3p21 (5), which encode the cardiac delayed rectifier potassium channel (IKr) and sodium channel (INa), respectively (Table 1⇓). These discoveries marked the start of an exciting and ever-expanding field of genetics of cardiac arrhythmias. A few months later, the first positionally cloned LQTS gene, KvLQT1 (KCNQ1), was reported (6), which when coexpressed with KCNE1 [encoding MinK (miniature K), a putative β subunit that exerts control of K+ channel gating] produces the slowly activating potassium current (IKs). Soon after, several KCNE1 mutations were reported in LQTS families (LQT5) (7, 8). LQT6 was identified as KCNE2 (9), a gene sharing significant sequence identity to KCNE1 and encoding a small β -subunit (MiRP1) that coassembles with KCNH2 to generate IKr (Figure 2⇓). Heterozygous mutations in KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 are associated with autosomal-dominant LQTS, whereas patients who possess homozygous or compound heterozygous mutations in KCNQ1 and KCNE1 have autosomal recessive LQTS with deafness (7, 10). The mutations identified in these genes produce an excess of late inward sodium current or reduced outward potassium current (11–13), both of which result in the prolongation of the ventricular action potential and the QT interval.
LQT4 was mapped to chromosome 4q25–27 in a large French family with autosomal dominant LQTS associated with sinus node dysfunction (bradycardia) and atrial fibrillation (14). Cloning of the LQT4 gene has remained elusive, but the recent identification of a loss-of-function mutation in ankyrin-B by Mohler et al. (15) finally puts to rest the search for the gene for LQT4 and establishes the ankyrin-B gene as the first non-ion channel gene involved in LQTS, providing a glimpse into new mechanism for the pathogenesis of LQTS.
Whereas ion channels are thought of as protein assemblies that exist in lipid membranes functioning, interacting, and coordinating with accessory proteins, ankyrins are a family of adapter proteins that link integral membrane proteins to spectrin-based cytoskeleton (16). Three members of this family have been identified as erythrocyte ankyrin (ankyrin-1 or ankyrin-R), brain ankyrin (ankyrin-2 or ankyrin-B), and general or epithelial ankyrin (ankyrin-3 or ankyrin-G). The ankyrin-B has three major isoforms with molecular weights of 440 kDa, 220 kDa, and 150 kDa, generated by alternative splicing. The major form of ankyrin-B in cardiac cells is 220 kDa. Ankyrins typically contain three functional domains that consist of the membrane binding domain, the spectrin binding domain, and the regulatory domain (16). Ankyrins bind to several ion channel proteins, such as the anion exchanger (Cl−/HCO3− exchanger), Na+,K+ –ATPase, voltage-sensitive sodium channel (INa), and Na+/Ca2+ exchanger (NCX or INa-Ca), and calcium-release channels including those mediated by the receptors for inositol trisphosphate (IP3) or ryanodine (16).
The recent study by Mohler and colleagues challenges the current direction of LQTS genetic research and looks beyond genes that code for ion channel proteins. The authors used both human genetics and engineered mouse models to establish that mutation E1425G in ankyrin-B on chromosome 4q25–27 is responsible for LQT4. This mutation was shown to cosegregate with LQTS and sinus node dysfunction in the patients, but not with normal family members in one LQTS family nor in control subjects. Using an elegant complementation test, the authors determined that E1425G was a functional mutation and not a rare polymorphism. Transfection of cardiomyocytes from heterozygous AnkB+/− mice with wild type ankyrin-B rescued the functional defects (abnormal Ca2+ dynamics) of these cells. However, mutant ankyrin-B with E1425G failed in this rescue, indicating that E1425G is a functional mutation that may act by a loss-of-function mechanism. Together with the finding that AnkB+/− mice displayed prolonged QTc (QT interval corrected for heart rate), sinus bradycardia, and ventricular arrhythmias induced by exercise or epinephrine, these data provide convincing evidence that ankyrin-B is the long-sought LQT4 gene. Nevertheless, identification of additional ankyrin-B mutations in other LQTS families or patients is needed to validate this exciting finding and to determine the frequency of ankyrin-B mutations in the LQTS population.
How does loss of ankyrin-B function cause ventricular arrhythmias? Mohler et al. examined the proteins that interact with ankyrin-B and found that expression of the α 1, α 2 subunits of Na+,K+ –ATPase, NCX (INa-Ca), and InsP3 R (inositol-1,4,5-triphosphate receptors) were reduced in adult AnkB+/− murine myocytes. The combined reduction in the expression of these and other yet to be identified ankyrin-B–interacting proteins may trigger the abnormal electrophysiological and Ca2+ dynamics in AnkB+/− cardiomyocytes. By way of Ca2+ transient measurements in isolated myocytes, Mohler et al. reported elevations in total [Ca2+]i. Cardiac β -adrenergic receptor activation, such as that caused by physical exertion or emotional stress, increases the Ca2+ influx into the cell and the intracellular demands for Ca2+ handling (Figure 3⇓). Activation of the ryanodine receptor pathway leads to localized sarcoplasmic reticulum (SR) Ca2+ release and to a cascade of events involving the INa-Ca current that regulates and maintains the pacemaker activity of the sinus node (17) (Figure 3⇓). If a deficiency in ankyrin-B expression disrupts [Ca2+]i homeostasis by causing the abnormal localization/organization of ion exchangers, then alterations in Ca2+ handling could partly account for the early and delayed after-depolarizations—EADs and DADs, respectively, a condition where a normal cardiac action potential is interrupted or followed by an abnormal early or delayed depolarization—in ventricular myocytes and the sinus node dysfunction observed by Mohler et al. in AnkB+/− mice.
However, the evidence for the QT interval prolongation and the abnormal slowing of conduction in adult AnkB+/− mice as functions of altered Ca2+ handling is unclear. In fact, human mutations in other Ca2+ handling systems such as in the ryanodine receptor 2 gene (hRyR2 ; resulting in increased sensitivity of Ca2+ -induced SR Ca2+ release) (18) or calsequestrin 2 gene (CASQ2 ; resulting in impairment of SR Ca2+ release) (19) manifested, respectively, normal to mildly prolonged QTc intervals (Table 1⇓, Figure 3⇓). Moreover, indications that elevations in [Ca2+]i may result in significant sinus node abnormalities or conduction defects were not found in the same patients with mutations in hRyR2 (18) who had normal sinus rhythm and PR interval (Figure 1⇓), or in CASQ2 patients (19) who exhibited resting bradycardia (Table 1⇓). Although Mohler et al. investigated the possibility of the effects of the ankyrin-B mutant on the L-type Ca2+ channel (ICa,L), no changes in this current were found in adult AnkB+/− mice. Interestingly, neonatal AnkB−/− and, to a lesser degree, AnkB+/− mice reported in a previous study (20) revealed abnormal cardiac Na+ channel kinetics. Although the impact of ankyrin-B deficiency expressed in the heterozygous phenotype of mice (and humans) is more subtle than those observed in the knockout mouse, the changes in Na+ channel kinetics in the heterozygous phenotype reported by Chauhan et al. (20) are evident nonetheless. The evidence, thus far, suggests that cytoskeleton-dependent modulation of ion channel function (by controlling channel kinetics or the number of functional channels at the plasma membrane) should have a significant contribution to the overall excitability of the cell. It is also likely that disruption in the final cytoskeletal framework of one or more integral proteins may lead to electrophysiological remodeling of other ion channels—this also remains to be elucidated. The effects of ankyrin expression on cardiac ion channels (particularly in an adult cellular cytoskeleton framework) require further investigation; many protein structures require a solid anchor and proper localization to convey the dynamic and precise mechanisms of excitable cells.
Strategies for clinical interventions are best formulated when based on an adequate understanding of the causes of disorders. Traditional antiarrhythmic therapy involves ion channel blockers and/or electrical interventions that control the rhythmicity of the heart. The identification of previous LQTS genes has led to ion channel-specific and trial-based therapies for LQTS patients. The sodium channel blockers mexiletine (21) and flecainide (22) can shorten QTc for LQT3 patients. Elevating the concentration of potassium in serum (23) and keeping potassium channels open longer may be beneficial for patients with potassium channel mutations (24). The work of Mohler and colleagues associates LQT4 to a mutation that expresses a non-ion channel protein. Therefore, does one treat the symptoms (i.e., abnormal Ca2+ handling) or the underlying disease (via gene-specific therapy)? In any case, what would be the most appropriate clinical intervention, particularly when it is not certain if the sinus node dysfunction, the prolonged QT interval, or both are direct predispositions to the arrhythmia? As part of LQTS treatment, blockade of β -adrenergic receptors represents the first choice for therapy (25) but this choice is contraindicated under conditions of excessive bradycardia that is likely to favor EAD-induced TdP (e.g., LQT3). Antiadrenergic therapies for LQT4 patients may be warranted to reduce the cellular demands for Ca2+ handling (Figure 3⇓). Although the mechanisms involving the ionic regulation of sinus node dysfunction remain unclear, mutations associated with INa (26) and ICa,L (27, 28) have been implicated as critical players, and the mechanism by which the ryanodine receptor controls Ca2+ release from the SR (17) might provide future insights into the clinical management of LQT4. The intriguing work of Mohler and colleagues has undoubtedly provided an alternative avenue from antiarrhythimic drug research and emphasized the research for intracellular signaling-related therapies associated with cytoskeletal proteins as alternative treatments of LQTS and related arrhythmias.
Summary of Genes Implicated in LQTS and Other Cardiac Arrhythmiasa
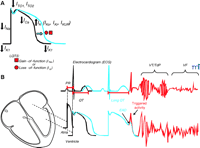
Cardiac action potential, electrocardiogram (ECG), and long QT syndrome (LQTS). A. In LQTS, mutations cause either a gain- or a loss-of-function in INa or IK currents, respectively, that results in prolongation of the cardiac action potential. B. Representative ECG traces in relation to normal atrial and ventricular action potentials. PR and QT intervals, respectively, correspond to the atrial and ventricular activation and repolarization times. Prolongation of the cardiac action potential is reflected as lengthening of the QT interval (blue), an increased vulnerability for the development of early afterdepolarization (EAD), “triggered activity,” and ultimately cardiac arrhythmias [torsade de pointes (TdP) or ventricular fibrillation (VF)]. IKr, rapidly activating delayed-rectifier potassium channel current; IKUR, ultrarapid delayed rectifier potassium current; ITO, transient outward potassium channel current; IK1, inward rectifier potassium channel current.
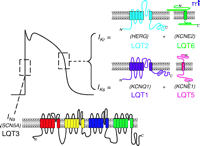
Cardiac ion channel subunits involved in LQTS. Topological maps of the voltage-gated α-subunit for INa (SCN5A) and α/β-subunits that make up IKr (HERG–KCNE2) and IKs (KCNQ1–KCNE1) are shown in relation to a typical ventricular action potential. Genetic mutations in these subunits are responsible for types 1, 2, 3, 5, and 6 of LQTS.

Candidate mechanisms for intracellular Ca2+handling in ventricular myocytes. The proposed mechanisms are as follows: (1) β-adrenergic receptor (β-AR) stimulation, via Gs-mediated activation of adenylate cyclase (AC) and protein kinase A (PKA), phosphorylates phospholamban (PLB) to relieve PLB’s inhibition of the Ca2+-ATPase pump (SERCA) and, thereby, to enhance Ca2+ uptake by the sarcoplasmic reticulum (SR). (2) Ca2+-induced Ca2+-release (CICR) through the ryanodine receptor (RyR) is mediated by L-(or T-) type Ca2+ channel activation. (3) CICR via activation of voltage-dependent INa and the dual roles of the Na+/Ca2+ exchanger (NCX). (4) Ankyrin-mediated (blue spheres) localization of ion transport proteins via spectrin binding. (5) Ca2+ release triggered by inositol -1,4,5-trisphosphate (IP3) through IP3 receptors (IP3R). (6) Inward rectifying K+ current (IK1) maintains resting membrane potential and is sensitive to changes in [Ca2+]i homeostasis. (7) CICR triggered by Ca2+ entry through tetrodotoxin (TTX)-sensitive Ca2+ channel. (8) Ca2+ release mediated by “slip mode conductance”, in which PKA-dependent phosphorylation of TTX-sensitive INa have altered permeability for Ca2+. (9) Ca2+-insensitve (ITO,Ca) and Ca2+-sensitive (ICl,Ca) transient outward potassium channels, the latter of which is carried by Cl− rather than K+ ions. (10) Reduced Ca2+-handling proteins due to a deficiency of ankyrin (blue spheres).
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Sandro Yong, PhD, (left) is a Postdoctoral Fellow in the laboratory of Dr. Wang in the Department of Molecular Cardiology, Lerner Research Institute and Center for Cardiovascular Genetics, Department of Cardiovascular Medicine at the Cleveland Clinic Foundation. Qing Wang, PhD, (center) is Associate Staff/Associate Professor of Molecular Genetics, Molecular Cardiology, and Cardiovascular Medicine and Director of Center for Cardiovascular Genetics at the Cleveland Clinic Foundation. QW is supported by NIH grants R01 HL65630 and HL66251, and a Doris Duke Innovation in Clinical Research Award. Please address correspondence to QW. E-mail wangq2{at}ccf.org; fax 216-444-2682. Xiaoli Tian, PhD, (right) is a Postdoctoral Fellow in the Wang lab Department of Molecular Cardiology, Lerner Research Institute and Center for Cardiovascular Genetics, Department of Cardiovascular Medicine at the Cleveland Clinic Foundation. XL is supported by an American Heart Association Ohio-Valley Postdoctoral Fellowship.

