Cytochromes P450, Drugs, and Diseases
Abstract
The human genome encodes fifty-seven cytochrome P45O (P45O, or CYP) proteins. The majority of these are involved in the metabolism of steroids, bile acids, fatty acids, eicosanoids, and fat-soluble vitamins. Nearly fifteen P45Os are involved in the metabolism of drugs and other xenobiotic chemicals and have received the most attention from pharmacologists. Many issues exist as to how to most logically deal with the major human P45Os in the context of the drug development process. Many of these same xenobioticmetabolizing P45Os also activate carcinogens, but the in vivo significance of such activation in cancer etiology has been difficult to assess because of the complexities of tumorigenesis. The functions of the remaining fifteen P45O "orphans" are unknown and represent an area in need of much study.1

Introduction
Cytochrome P450 enzyme (P450, or CYP) reactions were first recognized in the oxidation of drugs, carcinogens, and steroids,
and generally show the mixed-function oxidase stoichiometry: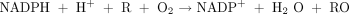
 In mammals, all P450s are membrane bound, a fact that hindered early studies. Most are found in the endoplasmic reticulum,
but five are localized primarily in mitochondria. However, work by Avadhani’s group has revealed that significant fractions
of several of the P450s that are usually considered to be microsomal can also localize to mitochondria (1). The P450s in the endoplasmic reticulum all interact with and receive electrons from a single flavoprotein, NADPH–P450 reductase.
The mitochondrial P450s use an electron transport chain with the iron–sulfur protein adrenodoxin and the flavoprotein adrenodoxin
reductase.
In mammals, all P450s are membrane bound, a fact that hindered early studies. Most are found in the endoplasmic reticulum,
but five are localized primarily in mitochondria. However, work by Avadhani’s group has revealed that significant fractions
of several of the P450s that are usually considered to be microsomal can also localize to mitochondria (1). The P450s in the endoplasmic reticulum all interact with and receive electrons from a single flavoprotein, NADPH–P450 reductase.
The mitochondrial P450s use an electron transport chain with the iron–sulfur protein adrenodoxin and the flavoprotein adrenodoxin
reductase.
The human genome encodes fifty-seven P450 proteins (2) (http://drnelson.utmem.edu/CytochromeP450.html). A recent survey (3) classified fifteen P450s involved in the metabolism of xenobiotic chemicals (i.e., chemicals, such as drugs, not normally found in the body): fourteen primarily involved in the metabolism of sterols (including bile acids); four that oxidize fat-soluble vitamins; and nine involved in the metabolism of fatty acids and eicosanoids. Substrates (either xenobiotic and endobiotic) are essentially unknown for the remaining fifteen of the fifty-seven. P450s are found throughout the phylogenetic spectrum: three have been identified in Saccharomyces cerevisiae, eighteen in Streptomyces coelicolor, eighty in Caenorhabiditis elegans, 257 in Arabidopsis thaliana, and perhaps surprisingly, none in Escherichia coli or Salmonella typhimurium.
In humans, the expression of some of the P450s is highly regulated but levels of others vary considerably. For instance, one plot of the variability of in vivo activity of CYP2D6 (5, 6) is shown in Figure 1⇓. Some individuals do not express CYP2D6 mRNA or protein, and some pharmacokinetic parameters can vary by a factor of 104 (e.g., metabolic ratio; see Figure 1⇓).
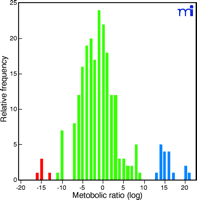
Relative rates of CYP2D6 activity in a Caucasian population (4). CYP2D6 activity is represented as the ratio of unmetabolized drug (here, debrisoquine) to drug metabolite (4-hydroxydebrisoquine) collected from the urine following a single dose. The distribution is the result of the existence of more than seventy alleles in the population (see http://www.imm.ki.se/CYPalleles/). Early research labeled individuals as “extensive metabolizers” (green) and “poor metabolizers” (blue) (5). The “ultrarapid metabolizer” phenotype (red) can result from inherited gene amplification (6).
The effects of missing a P450 vary considerably. Many of the P450s involved in the processing of steroids or vitamins, hence, a congenital defect may be very debilitating (7, 8), whereas, with other P450s, there is often little, if any, apparent phenotype associated with over- or underexpression. Transgenic mice apparently lacking orthologous genes corresponding to human “xenobiotic-metabolizing” P450s are generally normal physiologically (9). However, the expression of P450s can remarkably influence the therapeutic effectiveness and side effects of some drugs.
P450s as Therapeutic Targets
Some P450s are well-established targets for rational drug design. Chief among these is CYP19A1, the steroid aromatase. This P450 catalyzes the three-step oxidation of androgens to estrogens; decreased expression of CYP19 is desirable in estrogen-dependent tumors (10). Another long-standing target is CYP5A1, usually known as thromboxane synthase (11).
Other P450s are less well developed as targets. One possibility for further study is CYP3A4, the main human P450 in liver and small intestine. Research has been undertaken to find safe and effective inhibitors of CYP3A4 in order to enhance the bioavailability of expensive drugs such as HIV protease inhibitors. One obstacle to greater availability of P450 inhibitors is the general reluctance of the FDA and other regulatory agencies to approve mixtures of P450-inhibiting drugs without extensive testing. However, existing drugs have been used in auxiliary capacities. For example, the Australian Medicines Handbook notes that ketoconazole and diltiazem have been used as cyclosporin-sparing agents. Another set of theoretical targets for inhibition includes the P450s that produce biologically active oxidized eicosanoids (e.g., 20-hydroxyarachidonic acid and various arachidonic acid epoxides) (12, 13). However, most of these studies are still in their infancy, and the application of P450-inhibiting drugs in humans is not yet a reality.
P450s and Their Endogenous Substrates
The majority of the P450s with known functions are those that oxidize steroids and vitamins. Historically, less attention has been given to these P450s by pharmacologists working in drug metabolism, probably because of the relatively narrow range of substrate selectivity among these P450s. However, many of the reactions catalyzed by these P450s are well known to endocrinologists and those working with congenital disorders, and the field has been covered in a recent review (7). Most of our information on the molecular level has developed in the last five to ten years.
Associated with defects in at least thirteen different human P450s are several diseases, including glaucoma (CYP1B1), adrenal hyperplasia (CYP11A1, CYP21A2), mineralocorticoid excess (CYP17A1), and rickets (CYP27B1) (7). The endogenous P450 substrates may be divided into several major classes (see Table 1⇓): cholesterol and bile acids; steroids; prostaglandins; vitamins A and D; and other eicosanoids.
Endogenous Subtrates of P450s
P450s in these five categories generally do not contribute to the metabolism of drugs. However, CYP5A1 and CYP19A1 are already drug targets and others may become targets. The fungal analog of CYP51 is a target of azoles, and drug discovery is now focusing on differential inhibition of the fungal and human enzymes (18). Because various diseases arise from deficiencies of P450s, gene therapy may be of interest in the future.
P450s in Drug Metabolism
Almost twenty-five years ago, our laboratory embarked on the characterization of human liver P450s involved in the metabolism of xenobiotics, particularly drugs and carcinogens (19). Today, through the efforts of many laboratories, much is known about the functions of the P450s (Figure 2⇓), and non-invasive techniques have been developed to estimate their expression levels in vivo. Considerable information is also available about substrates, inhibitors, and inducers. Of the human genes coding for P450s, only five account for 95% of drug metabolism (Figure 3⇓). This accounting is not precise and changes as one moves from the liver to consider intestinal and other extrahepatic metabolism, and the paradigm of which P450s are most important may shift with time because most pharmaceutical companies have a bias against developing drugs that prove to be substrates for the highly polymorphic enzymes (e.g., CYP2C19 and CYP2D6). The dominance of five P450s in drug metabolism should not be surprising, because approximately fifteen of the fifty-seven P450s are devoted to the metabolism of xenobiotics, and also because they manifest high expression levels and broad substrate selectivity.
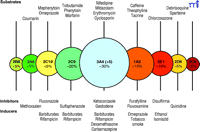
Major hepatic P450 enzymes involved in drug metabolism. Circles are intended to reflect the mean size of the pool of each of the main P450s (CYPs) in human liver (20–24). The exact pattern will vary among individuals. A few commonly recognized substrates, inhibitors, and inducers of these P450s are indicated. [ From an approach by Breimer. For extensive lists see Rendic (22).]
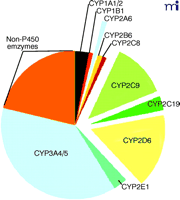
Contribution of major human P450s to the Phase I metabolism of all drugs currently marketed (25). The compilation is intended to be approximate and will vary with changes in drugs. For tables of raw data see Rendic (22). The non-P450 enzymes include alcohol and aldehyde dehydrogenases, flavin-containing monooxygenases, etc.
The state of knowledge about human P450s has advanced to the point where the FDA typically expects for the chemical entity in each New Drug Application a statement concerning its relationship to the P450s (e.g., substrate, inhibitor, and inducer of specific P450s and possibly other enzymes). One of the major concerns is avoiding drug interactions. This is an issue whose importance increases with the aging of the population (i.e., elderly people use more drugs). CYP2D6 has not been identified as having any “physiological” substrates—although some candidates have been considered (26, 27) —and individuals can tolerate wide variations (see Figure 1⇑) owing to the more than eighty known alleles in the population (5) (http://www.imm.ki.se/CypAlleles/). However, CYP2D6 “poor metabolizers” are at considerable risk when they encounter certain drugs (Figure 4⇓), as first observed in the study of Smith with debrisoquine (28, 29).
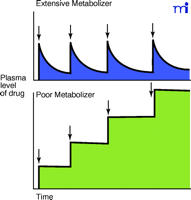
Significance of unexpectedly low metabolism of a drug by P450s. The typical pattern seen with the majority of the population (extensive metabolizers) is shown in the upper panel, where the plasma level of the drug is maintained in a certain range over the period of several consecutive doses (arrows indicate multiple doses). Unusually slow metabolism (lower panel) occurs when a poor metabolizer (without previous knowledge of phenotype) receives the same dose, resulting in an elevated plasma level of the drug. Adapted from (30).
Historically, drugs were developed with a relatively small number of individuals in trials, and doses were adjusted so that in most individuals the plasma level of drug (and presumably the level in the target tissue) would be maintained within a set range. However, in poor metabolizers, the same dose could yield a sustained plasma concentration and subsequent doses would increase this even more. Poor metabolism is especially problematic with drugs that have a narrow therapeutic index like debrisoquine (28), phenformin (31), or captopril (32). Although this phenomenon is now generally appreciated, there are still issues with older drugs (and possibly some newer ones that have escaped notice). For instance, Wedlund has estimated that psychiatric patients with CYP2D6 deficiency encounter 44% adverse drug incidents compared to 20% adverse drug incidents for extensive metabolizers (33). The cost of genotyping many individuals is considerably less than for a single serious incident. A case for using genotyping to achieve a higher success ratio with a drug has also been made in the case of haloperidol by Brockmoller and Roots (34).
Variability in pharmacokinetics can be an issue with other P450s. Several notable examples have been reported in the CYP2C subfamily. CYP2C19 is highly polymorphic, with variations in both the expression of mRNA and enzyme, plus actual differences in gene sequence (in the protein coding region) that give rise to differing rates of catalysis (35). As with most polymorphisms, there appear to be differences in expression in different ethnic groups. For example, the frequency of poor metabolizers among Asians is nearly 20% (cf ∼ 2% for Caucasians) (36). The proton pump inhibitor omeprazole and related ulcer drugs are oxidized by CYP2C19, and poor metabolizers show a better response to these drugs (37, 38). CYP2C9 also metabolizes (R )-warfarin. The therapeutic window is relatively narrow for balancing warfarin’s anticoagulant effect vs. hemorrhaging; fortunately, doses can be titrated in many cases, and the optimal dose of warfarin is known to depend on the presence or absence of the I359L polymorphism (39, 40).
One of the problems associated with using genotypes to predict pharmacokinetics and drug–drug interactions is that coding region substitutions may have varying effects in different reactions. For instance, the CYP2C9 I359L polymorphism affects the 6-hydroxylation of (R )-warfarin and the oxidation of the carbinol moiety on the antihypertensive prodrug losartan but not the 4′-hydroxylation of diclofenac (41–43). Of course, polymorphisms that affect enzyme levels, in contrast, affect all reactions of the enzyme.
Not all drugs interactions are genetically determined. In some cases, an inhibitor can block metabolism of a drug and produce the same effect as would poor metabolism. For instance, terfenadine, the first non-sedating antihistamine on the market, is essentially a prodrug, being rapidly oxidized by CYP3A4 (44). The drug was widely used with safety, but a few individuals experienced serious arrhythmias and died, with terfenadine accumulating in their plasma (45). Following this knowledge, the package insert contained a warning about contraindication for use with the CYP3A4 inhibitors ketoconazole and erythromycin. Eventually, terfenadine (i.e., the prodrug) was removed from the market and replaced by its active metabolite fexofenadine. In retrospect, terfenadine was a relatively successful drug in terms of the limited problems associated with extremely wide use, the deaths of some individuals notwithstanding. However, some of the problems could have been predicted with the use of the screening programs currently in place in most companies (i.e., in vitro “metabolic stability” assays). For example, many drugs are now packaged with a warning against their use following grapefruit ingestion. Such warnings arose from a serendipitous control experiment in a study of the interaction of ethanol with the calcium ion blocker felodipine (46, 47), whereby an active principle in grapefruit (i.e., bergamottin) proved to inactivate intestinal CYP3A4. Standard metabolic stability assays have subsequently established potential increases in exposure (i.e., area under the curve) to a variety of drugs that could prove dangerous if ingested along with components of grapefruit.
Unusually high amounts of P450s also can be problematic and may originate from gene duplication (6) —as in the case of “ultra” rapid metabolizers—or, more commonly, arise from enzyme induction. A classic example involves CYP3A4 and 17α -ethynylestradiol, the estrogenic component of oral contraceptives. Reports of oral contraceptive ineffectiveness following ingestion of rifampicin or barbiturates (48) were followed by demonstration of the accelerated clearance of 17α -ethynylestradiol (49) involving CYP3A4 (50). Similarly, hyperforin, a potent P450 inducer found in the herbal medicine St. John’s wort (51, 52), greatly increases the expression of P450s that metabolize drugs used for AIDS treatment (53) and organ transplantion (54).
Cases for enhanced drug toxicity due to elevated levels of P450s are probably less clear; however, CYP3A4 does convert the antidiabetic drug troglitazone into toxic products (55), although the mechanism of toxicity is still unclear. Troglitazone has since been removed from the market. In any event, the drug development process now incorporates a variety of in vitro studies designed to predict bioavailability, inhibition of P450 reaction, and the effects of any induction prior to consideration of clinical trials. Tucker has developed software to transform in vitro results with a drug to in vivo predictions regarding population pharmacokinetics (56).
In principle, genotyping can provide useful information about the expected behavior of a drug. However, large-scale single nucleotide polymorphism (SNP) analyses of P450s have not been done for many clinical trials. One issue is the cost associated with a large-scale SNP analysis of the many genes associated with transport, metabolism, and receptor antagonism, etc. Another issue is the limited amount of information available about most of the identified SNPs. The alleles most frequently associated with aberrant mRNA splicing in the CYP2D6 and CYP2C19 polymorphisms are known, but little functional information is available (e.g., www.imm.ki.se/CYPalleles/) for most of the identified SNPs. Studies of coding region variants of human P450s have revealed relatively small differences in catalytic activities (57), although there has been a tendency to overemphasize some of these differences. Predictions based upon homology modeling of variations in the catalytic activity of these P450 variants have not been generally useful. Furthermore, some P450 variants show preferences for substrates unlike that of the wild-type enzyme (58). Recently, we expressed the four known coding-region allelic variants of human CYP1A2; one did not express well but the other three exhibited only ∼ 2-fold variation in kcat /Km or kcat compared to the wild-type allele for most assays (H. Zhou, D. Kim, F. P. Guengerich, and P. D. Josephy, in preparation).
P450s and Cancer Risk
Two of the driving influences in P450 research have been the ability of P450s to activate procarcinogens (59) and the desire to exploit the regulation of individual P450s and their variability for purposes of cancer prevention and treatment (60). Experimental models have clearly demonstrated that the modulation of P450 expression can modify the susceptibility of animals to cancers produced by various chemicals (61, 62). The relevance of P450 modulation to cancer risk has not been easy to establish in humans, however. Nonetheless, in vitro studies have largely established that human P450s can activate most major chemical carcinogens (30, 63, 64). A diagram similar to that shown in Figure 3⇑ can be constructed, with some differences. The main P450s involved in carcinogen activation appear to be CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2E1, and CYP3A4, with some contributions from CYP4B1 (65) and CYP2A13 also possible (66, 67).
Epidemiological studies regarding P450 variants and cancer risk are unclear. Early research suggested that the risk of lung cancer in smokers was associated with the inducibility of benzo[a ]pyrene 3′-hydroxylation by CYP1A1 and possibly CYP1B1 in peripheral blood lymphocytes (68). These results have been difficult to reproduce and their significance and mechanism still remain uncertain. Another set of studies on lung cancer investigated CYP2D6 and a possible reduced risk in smokers with the poor metabolizer phenotype (69). However, no carcinogens have been identified that are preferentially activated by CYP2D6 (70), which is poorly expressed in lung, and further epidemiology/meta-analysis has not substantiated the original results (71). Numerous attempts have been made to link CYP2E1, known to activate many carcinogens (72), with cancers (73, 74) ; however, few, if any, of the SNPs analyzed with non-invasive probes (e.g. chlorzoxazone 6-hydroxylation) revealed in vivo differences in activity (75). Similarly, a suspected association between CYP3A4 and prostate cancer (76) has not been reproducible (77–79). Another problem with many of the epidemiological studies is that exposure to alleged substrates of the P450s has been presumed without proof. We can contrast the difficulty in the epidemiology of cancer etiology with drug clinical trials. Clinical trials often involve the careful analysis of the fate of a defined dose of a single drug in thousands of individuals, and yet the importance of a P450 genotype may still not be very obvious. Difficulties in studying the association of cancers with toxic chemicals in the environment are made more complex by the usually very limited numbers of patients available, the need to identify individual chemicals and levels of exposure, and the long latency period (up to twenty years) from exposure to onset of cancer.
What are the most likely P450s involved in cancer? Some of the best current candidates are CYP1A2 and CYP2A6. CYP1A2 activates many heterocyclic amines present in pyrrolyzed food, especially charred meat. Epidemiological studies have shown increased risk of colon cancer in individuals with high CYP1A2 activity (as judged by measurement of caffeine metabolism) but only when coupled with the rapid N -acetyltransferase phenotype and high consumption of “well-done” meat (80). CYP2A6 activates some nitrosamines and is expressed in the head and neck; a cancer risk may thus be associated with higher expression of CYP2A6 (67). A study by Kamataki and colleagues associated a poor metabolizer genotype with decreased risk of lung cancer in Japan (81). One factor in the analysis, however, may be a tendency for smokers deficient in CYP2A6 to smoke less because of its involvement in nicotine metabolism (82).
Two other candidates possibly connected with cancer include CYP1B1 and CYP2A13. CYP1B1 is expressed in several organs and tissues (e.g., breast, prostate, and ovary) and activates numerous carcinogens in vitro (83). The estrogen 4-hydroxylation activity of CYP1B1 is of particular interest with regard to estrogen-responsive tumors (84, 85). Allelic variants of CYP1B1 are known but these do not seem to differ markedly in regard to their catalytic activities (86), with the exception of the premature protein truncation that is associated with a hereditary glaucoma (87). Lastly, CYP2A13, which is similar to CYP2A6, activates nitrosamines and is localized in tissues of the respiratory tract (66, 67).
Some Future Directions in Human P450 Research
P450s will continue to receive intense attention because of their ability to metabolize drugs and endogenous compounds such as steroids, eicosanoids, and fat-soluble vitamins. Investigation into P450 activities must be accompanied by an appreciation of transporters and conjugating enzymes, systems not treated here in the interest of brevity.
What will be the likely future of P450 research? One consideration is how much screening will be done in preclinical drug discovery and development; however, there are valid questions about how the screening process influences the time to market and the cost of drug development. Perhaps a better question is how we can make the in vitro screening phase more useful. The prospect of coupling a massive scale SNP program with clinical trials has been considered, but such efforts are quite limited to date. The outlook would probably change if multi-SNP analysis led to the successful development of a useful drug. SNP analysis of P450s would probably be most effective in the selection of drugs that can be tolerated by all genotypes, rather than the multiple-track development of drugs to fit individual genotypes. (The latter approach would multiply the cost of drug development, safety assessment, and clinical trials, and retain liabilities for problems with drug metabolism.)
Another important issue is that of the “orphan” P450s for which little functional data exist. At least fifteen of the fifty-seven P450 gene products can be classified in this group; however, based upon our present knowledge, these P450s are unlikely to play significant roles in the metabolism of many drugs. These orphans might contribute to vitamin metabolism or possibly to the disposition of carcinogens in target tissues. Defining their sites of localization is possible in tissues, whereas identifying which cell types express them may be more problematic. Establishing the function of the orthologs of these genes may be possible with transgenic mice. However, identifying and characterizing the reactions catalyzed by orphan P450s is a challenge and there are opportunities for using new approaches. In much of the early work with animals, P450s were purified and then put through batteries of possible reactions (88). This approach had some success but most of the successful human P450 research has involved purifying P450s using assays involving known reactions and substrates (89). Neither approach will probably be very successful with the remaining orphans without more hints about possible ligands.
Research into P450s may have relevance to drug discovery, as well as to drug development. Some possibilities have been covered in a recent review (90). For instance, human P450s can be produced in heterologous expression systems and used to generate drug metabolites for more screening. Some current work in our laboratory involves the use of human P450s and several mutants [derived from “molecular breeding” (i.e., random mutageness, or directed evolution) (91) ] to generate novel indigo-like compounds with activity as protein kinase inhibitors (92). Prokaryotic P450s can also be considered as prospects for drug discovery and synthesis, and a proposal has been made to use Streptomyces P450s in the search for new antibiotics (86).
These are only a few aspects of the future of P450 research. Obviously, many more specific questions need to be addressed in both basic and applied research. Imagination, resourcefulness, and tenacity have been partners in the development of the P450 field and will continue to be in the future.
Acknowledgments
P450 research in the author’s laboratory has been funded in part by USPHS grants R01 CA90426 and P30 ES00267. Thanks are extended to J. Nelson for the invitation to contribute this review and to E.M.J. Gillam for her comments.
Footnotes
-
↵1 This review is the first in a series on cytochrome P450 enzymes.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

F. Peter Guengerich, PhD, is Professor of Biochemistry and Director of the Center in Molecular Toxicology at Vanderbilt University School of Medicine. His interests include the characterization of human P450s and mechanisms of P450 catalysis. He has been an ASPET member since 1979 and received the John J. Abel (1984) and Bernard B. Brodie (1992) Awards for his research. E-mail f.guengerich{at}vanderbilt.edu; fax (615) 322-3141.

