Epigenetic Downregulation of GABAergic Function in Schizophrenia: Potential for Pharmacological Intervention?
- Send correspondence to EC. E-mail costa{at}psych.uic.edu; fax 312-413-4569.
Abstract
Although the propensity to develop schizophrenia has long been attributed in part to a “genetic” component, this component does not follow Mendelian laws and may be primarily epigenetic. We have detected a decrease in levels of mRNA encoding the 67-kDa glutamate decarboxylase (GAD67), but not of that encoding GAD65, in several cortical areas from the brains of patients afflicted with either schizophrenia or bipolar disorder, but not from patients with severe depression. In addition, dendritic levels of reelin (and the mRNA that encodes it), a protein essential to neurodevelopment, are decreased in schizophrenia. We have further investigated whether hypermethylation of the genes that encode the GADs and reelin might be at the root of the observed decreases in corresponding mRNA. Indeed, administration of methionine, a source of methyl groups in multiple enzymatic reactions, causes a recrudescence of schizophrenic symptoms, and we have correlated high intake of methionine in mice with both an increase in levels of promoter methylation and a decrease in production of reelin and GAD67. This decrease is reversed by treatment with valproate, an inhibitor of histone deacetylase.
Introduction
Although several expansive, multigene loci have been linked to schizophrenia vulnerability, attempts to identify specific genes have been disappointing (1). In fact, nearly every chromosomal locus implicated through linkage analysis has evaded statistical validation as a genomic determinant of the disease (2, 3). Although positive signals from independent studies are beginning to converge on similar chromosomal locations, the regions so far identified are large and include a vast number of candidate genes, and so no specific gene can yet be targeted for investigation or therapy.
Can mRNA microarray technology help identify a genetic background reliably linked to schizophrenia vulnerability? Recently, Hakak et al. (4) found eighty-nine genes, of a total 6000 genes examined from the dorsolateral prefrontal cortex, to manifest transcriptional rates particular to schizophrenia, including genes operative in synaptic plasticity, neuronal development, neurotransmission, and signal transduction. Mirnics et al. examined 7000–8000 genes in the same brain region and ascribed to all schizophrenics studied defects related to presynaptic function, including regulator of G protein signaling (RGS) 4 (5–6). The RGS4 gene maps to chromosome 1q21–22, which coincides with an area implicated in schizophrenia by linkage studies.
The interpretation of available microarray data to identify specific molecular correlates of schizophrenia is complicated by several factors, not the least of which pertains to the unfortunate reliance on heterogeneous cell populations from cortical and subcortical brain regions. Recently, single-neuron microdissection of schizophrenic brain tissue, coupled with linear antisense RNA amplification of high-density gene arrays (18,000 genes), has implicated marked differences in G protein signaling, as well as in the expression of glutamate receptor subunits and synaptic proteins, in stellate neurons from entorhinal cortex layer II (7). Nevertheless, the abundance of various mRNAs whose genes map to sites presumably linked to schizophrenia remain largely unchanged by the disease (7).
Taken together, these findings may reflect epigenetic mechanisms of gene regulation and underscore the need for analyzing homogeneous collections of neuronal phenotypes. Indeed, carefully executed gene expression analysis will be crucial to accelerating the slow progress of genetic research into complex diseases that has typified linkage analysis (8). Genetic linkage studies are inherently oriented to the detection of disease-related DNA mutations and polymorphisms, which are operative, at most, in only a few sporadic cases of schizophrenia.
Schizophrenia: More Than Just the Sum of Multiple Genes
The 1982 publication of Gottesman and Shields (9) still gives an accurate appraisal of our understanding of the etiology of schizophrenia. In contrast, elaboration of the mechanisms attending the epigenetic regulation of gene expression has progressed dramatically over the past two decades. In fact, the term epigenetic indicated in 1982 vague theories based on abstract concepts of gene–environment interactions, whereas the word today implies well-defined chemical modifications that alter chromatin structure and gene transcription. The methylation of CpG islands within promoter sites (i.e., formation of 5-methylcytosine), catalyzed by a DNA cytosine-5 methyltransferase (DNMT), is a prime example (10, 11). (See Box.)
The widely observed phenotypic variability of human neuronal brain circuitry (34, 35) and the continuous remodeling of selected neuronal circuits in relation to learning and memory (36) resonate with the biological relevance of epigenetic transitions in gene funtion. Genomically, brain function is encoded similarly across species and is basically identical among individuals within species; however, the complexities of neuronal circuitry, including dendritic spine volume, shape, and turnover may differ radically as a result of individual experience that accesses, via afferent input, the brain’s functional and structural plasticity (36). Investigation of a number of epidemiologic features of schizophrenia thus requires an understanding of neuronal plasticity that is beyond the scope of traditional Mendelian genetics.
For example, the schizophrenia rate for monozygotic twins is fifty percent, even though they arise from identical DNA sequences. Significantly, analyses of genomic DNA from monozygotic twins discordant for schizophrenia have shown discrepancies in the methylation patterns of certain gene promoters (37). The level of DNA methylation within a given gene promoter, moreover, can vary not only between individuals, but also within an individual over time (38). Intriguingly, a fluctuating course is typical of schizophrenia, and so longitudinal studies to monitor the sites of DNA that undergo modification and demodification as the patient goes through remission and relapse may well help in identifying the epigenetic components of schizophrenia. The onset of schizophrenia symptomatology at puberty, for example, might be related to hormone-induced epigenetic changes in brain-specific promoters. It is well known that adrenal and sex hormones have an impact on gene expression via changes in higher order chromatin remodeling (39, 40). Epigenetic mechanisms related to the hypermethylation and concomitant downregulation of promoter function are not only specific to cell types (e.g., neurons) (41) , but can even be specific for cellular subtypes (see below).
Epigenetic Factors in Neurodevelopment Disorders
Can detailed knowledge of epigenetic mechanisms and their aberration in transformed (i.e., replicating) cells (see Box) be extended to neurons (i.e., postmitotic cells) and neurological disorders? The molecular biology of Rett syndrome, as well as of fragile X syndrome, indicates that aberrant epigenetic mechanisms involving MBDs (in association with DNMT, HDACs, and HMTases) result in inappropriate gene regulation (41). In particular, a neuronal deficit of the MBD protein MeCP2 has been implicated in Rett syndrome, whereby insufficient levels of HDACs are found in association with chromatin, so that histones remain hyperacetylated and promoters are concomitantly hypomethylated and overactive (42). In fragile X syndrome, on the other hand, hypermethylation (and underutilization) of the promoter region of a gene (FMR1 ) essential to dendritic spine plasticity appears to ensue from an unusually high CpG content (43, 44). Whether this syndrome would be amenable to treatment with HDAC inhibitors remains to be seen.
EPIGENETIC MECHANISMS OF REELIN AND GAD EXPRESSION IN SCHIZOPHRENIA
Indications that epigenetic regulatory mechanisms operate in the etiology of schizophrenia can be garnered from examination of the expression levels of at least three genes. In every structure of schizophrenic brains examined postmortem (i.e., frontal and parietal cortices, hippocampus, neostriatum, and cerebellum), the genes that encode reelin and GAD67 (RELN and GAD1 ), but not the gene that encodes GAD65 (GAD2 ), are downregulated at both the transcriptional and translational levels (45–48). The decrease in GAD67 and RELN expression is also typical of bipolar disorder with psychosis, but is not characteristic of patients with major depression without psychosis (47).
In contrast, postmortem examination of prefrontal cortex demonstrates that DNMT1 , which in healthy brains is preferentially expressed in GABAergic interneurons (Figure 1⇓), is further upregulated, specifically in these cells, in schizophrenia. Increases in DNMT1 mRNA are evident primarily in layers I, II, and IV. The increased production of DNMT1 in particular implies an epigenetic mechanism (i.e., the hypermethylation of CpG islands) for the downregulation of RELN and GAD1. Indeed, the promoters for both RELN and GAD1 are embedded within CpG islands, and we have shown that the expression of RELN can be significantly altered by interfering with the cellular epigenetic regulatory apparatus (49, 50). Furthermore, increased DNMT1 mRNA in cortical GABAergic neurons is consistent with the decreased utilization (i.e., presumably, through hypermethylation) of the RELN and GAD1 promoters (1, 51). Circumstantial in vivo evidence for the epigenetic downregulation of genes related to GABAergic function in schizophrenia is also provided by the promoter hypermethylation of Reln that is observed upon protracted treatment of methionine in mice. The identification of methylcytosine nucleotides in the RELN promoter of bisulfite-treated genomic DNA isolated from schizophrenia and nonpsychiatric patient brain samples (D. Grayson, unpublished results) should mandate investigation into the likelihood that other CpG island–rich promoters operative in cortical GABAergic neurons may be hypermethylated in schizophrenia.
EPIGENETIC REGULATION OF GENE EXPRESSION
Promoter Regulation by Methylation of CpG Islands
In mammals, the methylation of CpG islands within specific promoters (12), catalyzed by DNA cytosine-5 methyltransferases (DNMTs), is a common epigenetic modification. DNMTs catalyze the transfer of the methyl group from S -adenosylmethionine (SAM) to the 5 position of the cytosine of CpG dinucleotides. Computational analysis of the human genome sequence predicts the existence of 29 x103 CpG islands (13, 14). Whereas DNA hypermethylation at CpG islands is associated with transcriptionally inactive chromatin, hypomethylation is associated with transcriptionally active chromatin (15; see the Figure).
Compared to the genome at large, most promoter sequences enriched in CpG islands are undermethylated. However, a small but significant proportion of these CpG islands become completely methylated during development, so that the associated promoters remain silent. Accordingly, aberrant promoter methylation can be essential to certain cancers and neurodevelopmental disorders (e.g., schizophrenia).
Transcriptional Regulation of the DNMT1 Gene
Several lines of evidence support the notion that DNMT1 levels are transcriptionally regulated (16). For example, activation of the Ras–activator protein-1 (Ras–AP-1) signaling pathway induces DNMT1, the promoter of which has an AP-1 site nested within a CpG-rich area (17). Moreover, three c-Jun dependent enhancers are located downstream from transcription initiation, thus providing a molecular explanation for the responsiveness of DNMT1 to oncogene signals in cancer.
Covalent Modifications of the Nucleosome Histone Core
In eukaryotes, promoter DNA methylation is tightly regulated by high-order chromatin structure remodeling (see Figure). The structural components attendant to such chromatin remodeling include: i) histones, ii) chromatin remodeling proteins, and iii) methyl-CpG–binding domain proteins (MBDs). The best-studied MBD is MeCP2, a 486-residue protein abundantly associated with the chromatin of various neurons. MeCP2 binds to methylated CpG dinucleotides and represses transcription by recruiting HDAC in complex with other proteins. In experimental (transformed) cell types, inhibitors of HDAC activity, promoting histone hyperacetylation, result in the partial derepression of genes and concomitant release of MeCP2(18).
Chromatin remodeling involves the covalent modification (e.g., acetylation, methylation, phosphorylation or ubiquitination) of the eight N-terminal tails of the nucleosomal histone octamer (11, 19, 20). Several lines of evidence suggest that acetylation and methylation of histone tails are directly associated with DNA promoter methylation. The steady-state level of cellular histone acetylation is maintained by the opposing actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs)(21). Certain HAT complexes may include general transcription factors, and HDAC complexes can include transcriptional repressors (corepressors, e.g., Sin3).
In addition to acetylation or deacetylation, the methylation of histone N termini, catalyzed by the histone methyltransferases (HMTs), has been implicated in the chromatin remodeling machinery and thus in gene expression(11). There is, moreover, an interaction between histone tail methylation and acetylation (22–24). Histone acetylation is associated with hypomethylation of both DNA and histones. On the other hand, histone methylation seems to be connected with increased DNA methylation and conversion of euchromatin to inactive heterochromatin (25). Histone methylation presumably facilitates DNA methylation by recruiting heterochromatin protein 1 (HP1), which in turn recruits DNMTs to promoter sequences targeted for silencing (i.e., containing CpG islands) (11, 26, 27). In this way, histone methylation represents an important epigenetic signal operative in the higher-order chromatin remodeling that effects transcriptional downregulation (probably via CpG island hypermethylation).
MBD2/Demethylase Activity
Several laborious efforts have been made to isolate an enzyme activity that could reverse the methylation reactions catalyzed by DNMT (28). The most impressive catalytic activity to be discovered in human cell extracts (29) is associated with the MBD2 protein. Overexpression of MBD2 in vitro can reverse the methylation of some genes but not of others. The MBD2 protein is the only member of the MBD family reported to have both transcriptional repressor and DNA demethylase activities (30). Although it has been suggested that such duality of function may be needed to coordinate a concerted program of gene activation and repression, attempts by several laboratories to replicate evidence of a demethylase activity associated with MBD2 have not been successful (31). Nevertheless, the search for demethylating enzymes was reinvigorated upon demonstration that, following fertilization, the paternal genome undergoes extensive demethylation (32).
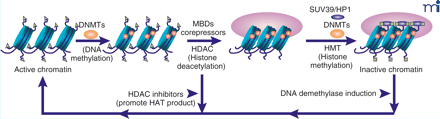
Chromatin remodeling processes. The cartoon depicts changes that occur as chromatin is remodled from active chromatin inactive chromatin. The transition is a multi-step process that involves: a) DNA methylation by DNA methyltransferases (DNMTs); b) binding of methyl CpG dinucleotides by MBD proteins; c) histone deacetylation by HDACs; d) HMTs (e.g., SUV39) that methylate histones and that are recruited by sequence-specific DNA binding proteins (e.g., HP1). HDAC inhibitors can reactivate gene transcription by increasing histone tail acetylation, which relaxes chromatin. The DNA demethylase activity of MBD2 may be involved in reversing the methylation of promoter regions, a first step in reversing gene silencing. MBD, methyl binding domain proteins; HDAC, histone deacetylase; HMT, histone methyltransferase; HP1, heterochromatin binding protein; DNMT, DNA methyl transferase. Green triangles represent the acetylation of histone tails. Green boxes represent the methylation of histones. Figure is modified from (33).
Cortical GABAergic Neurons: Production Centers of the Signaling Protein RELN
In the adult mammalian neocortex, RELN, comprising 3,461 amino acids, is synthesized and secreted by GABAergic cells into the extracellular matrix (ECM). The secreted protein is recognized by integrin receptors located on dendritic spine postsynaptic densities of pyramidal neurons (51–55) and may contribute to the regulation of neuropil expression and synaptic plasticity by modulating rates of spine turnover, morphology, and interactions with cortical afferent thalamic axons. Immunoelectron microscopy indicates that after its release into the ECM, RELN adheres to integrin receptors (α 3, β 1, or α 5,β 1) expressed in dendritic shafts and spines and thereby surrounds spine postsynaptic densities and leads to the cytosolic activation of the adapter protein Disabled-1 (Dab-1; Figure 2⇓) (49, 51–54, 56, 58–60). The signal transduction cascade that begins with the binding of RELN to integrin receptors may function ultimately to support synaptic plasticity through reorganization of the cytoskeleton.
The very selective expression of RELN by GABAergic neurons may occur in additional brain structures (e.g., medium spiny neurons of ventral striatum, but not Purkinje cells of cerebellum) throughout the life span of mammals (45, 54). Protein synthesis appears to be key to the sequence of events initiated by RELN and most likely functions to enhance LTP (61). It is believed that the translation of mRNAs resident in dendritic spines is activated by a complex signaling cascade leading to the phosphorylation of a cytoplasmic polyadenylation element binding factor (CPEB) that activates an mRNA polyadenylate polymerase, an event required prior to translation (62). It thus appears that RELN, as well as the mammalian target of rapamycin (mTOR), activates protein synthesis by facilitating the polyadenylation of dendritic spine mRNA.
RELN Downregulation and Neuronal Hypoplasticity in Schizophrenia
The dendritic spine density in cortical pyramidal neuron arborizations (layer III) is decreased in schizophrenia, suggesting a compromise in neural plasticity (63, 64). Postmortem evaluation of the brains from schizophrenic and bipolar patients cannot distinguish whether a neuropil plasticity deficit leads to the onset of GABAergic molecular peculiarities that include RELN and GAD1 downregulation or whether such downregulation is the primary cause of these two psychiatric disorders. Is reelin haploinsufficiency operative in schizophrenia vulnerability? Hong et al. (65) convincingly showed that an autosomal form of lissencephaly that is associated with severe abnormalities of the cerebellum, hippocampus, and brain stem maps to chromosome 7q22 and includes one of two independent RELN mutations known to severely curtail RELN protein biosynthesis. It is tempting to speculate that the lissencephaly associated with the reeler human phenotype is equivalent to the severe cortical neuronal disorganization of the null mutant reeler mouse, but humans who are heterozygous (RELN+/- ) exhibit a very severe mental deficiency that unfortunately precludes detection of the psychiatric symptoms necessary for a diagnosis of schizophrenia.
Glutamic Acid Deacarboxylase Activities: Regulatng Production of GABA
Two molecular forms of glutamic acid decarboxylases (GAD65 and GAD67 ) regulate the synthesis of GABA in the mammalian brain. They are encoded by two different genes, both expressed in GABAergic interneurons (66). The two enzymes differ in structure and function. GAD67 contains a pocket that covalently binds pyridoxal phosphate, whereas GAD65 is generally associated with the cofactor solely during catalysis. GAD67 activity is regulated at the level of gene expression, whereas GAD65 provides a basal rate of GABA production.
In knockout mice, the obliteration of GAD67 activity causes cleft palate, suggesting a role for this enzyme in craniofacial bone formation. Because of this malformation, homozygous GAD67 null mice cannot be used experimentally. The lack of GAD65 in knockout mice facilitates spontaneous and electrically induced convulsive activity. However, in the GAD65 and GAD67 heterozygous mice the expression of reelin is normal, whereas in the HRM, which express fifty percent of reelin, the expression of GAD67 but not GAD65 is also downregulated, although the molecular mechanism of the interaction between GAD67 and reelin mRNA expression has not been elucidated. Because the promoters of both GAD1 and GAD2 are embedded in CpG islands, and because the GAD1 gene transcription rate is much faster than that of GAD2 , a methylation that occurs in the CpG islands of the two GAD genes is more effective in decreasing the transcription of GAD1 , the gene that proceeds at a faster rate. The human GAD1 gene maps to chromosome 2q31 and GAD2 maps to chromosome 10p11.23. Linkage studies have failed to implicate these two loci as susceptibility factors in schizophrenia. The mechanism by which the expression of GAD1 (encoding GAD67 ) is selectively and consistently downregulated in bipolar disorder and schizophrenia is not yet clear.
Genetic Hypermethylation in Schizophrenia: Implications of Methionine Administration
It has been known for forty years that administration of methionine to schizophrenic patients results in a profound exacerbation of schizophrenia symptoms in sixty to seventy percent of patients (67–70). The administered methionine does not result in increased levels of methylated catecholamine metabolites and elicits no response from normal control subjects. In keeping with the epigenetic hypothesis of schizophrenia vulnerability, we must consider whether the exacerbation of the psychotic symptoms in schizophrenic patients is due to hypermethylation of CpG island gene promoters in GABAergic neurons, further lowering their already compromised levels of GABA and RELN function. Indeed, the protracted injection of methionine into mice increases brain S -adenosyl methionine (SAM) levels and decreases the expression of mRNA encoding reelin and GAD67 in the frontal cortex (Figure 3⇓). The effect of methionine was associated with an increase in the number of methylated cytosines in the Reln promoter present in the cortex (Figure 3⇓).
Prospective Pharmacological Regulation of RELN and GAD Hypermethylation
Based on circumstantial evidence that increased DNMT1 activity is associated with schizophrenic sympomatology, a theoretical strategy to study the effects of experimentally reduced hypermethylation of RELN or GAD1 promoters would be to inhibit DNMT activity in methionine-treated mice. Unfortunately, specific DNMT inhibitors are not available, and the use of specific antisense DNA sequences to the known DNMTs appears problematic due to the blood–brain barrier (10).
One of the regulatory mechanisms involved in the control of promoter gene methylation by DNMTs is this enzyme’s accessibility to DNA substrates. This accessibility depends on nucleosomal integrity and the acetylation status of histones, which is governed by the balance of HAT and HDAC activities. Studies carried out in the treatment of tumors suggest that the increased DNMT activity observed in tumor cells can be the result of an increased direct interaction of DNMT with HDACs (15). Hence, using methionine-treated mice as a model system, we have focused on the action of HDAC inhibitors, which may normalize possible epigenetic DNA methylation defects by increasing core histone tail acetylation at nucleosomal sites. Valproate, which inhibits HDAC activity, is widely prescribed as a mood stabilizer and to support antipsychotic therapies.
To establish whether valproate, in doses required to effectively potentiate the efficacy of antipsychotics in schizophrenia patients, completely inhibits brain HDAC activity in mice, we examined by western blot and immunochemistry the acetylation state of histone H3 and H4. We found that valproate produces a dose-related increase in acetylation of histone H3 in the frontal cortex. The increase in acetylation of histone H3 in interneurons normalizes the methionine-mediated downregulation of RELN and GAD1 (Figure 3⇓), suggesting that valproate may reduce GAD1 and RELN promoter hypermethylation by promoting nucleosomal acetylated histones in GABAergic interneurons. We suggest that a pharmacological intervention that normalizes Reln expression in the heterozygous reeler and in methionine-induced hypermethylation mouse models may be useful in developing treatments for schizophrenia.
Conclusions
Although inheritable factors appear to contribute to the etiology of schizophrenia, not a single gene mutation has been linked to schizophrenia vulnerability. Because the heritable component of schizophrenia does not follow inheritance predicted by Mendelian laws, an evaluation of possible epigenetic dysregulation of gene expression as a etiopathogenic mechanism of schizophrenia should be explored. Here, we have reviewed the principal arguments and new emerging facts in support of an epigenetic hypothesis of schizophrenia. The arguments that an investigation into the epigenetic regulation of genes may lead to a better understanding of the etiopathogenic mechanisms of schizophrenia vulnerability include: a) discordance of schizophrenia morbidity between monozygotic twins; b) late peri-pubertal age of onset at a time of hormonal changes; and c) sporadic schizophrenia cases that are incompatible with an inherited gene mutation. It is very likely that a study of the regulation of RELN and GAD1 expression in specific neuronal populations (i.e., selective GABAergic interneurons operative in cortex) may help to identify epigenetic molecular mechanisms operative in schizophrenia.
Understanding how epigenetic mechanisms can regulate RELN and GAD1 transcription is likely to open the way to the discovery of new drugs that can reprogram transcriptional programs that support neuropathology. Direct evidence that DNMT1 mRNA expression is increased in cortical GABAergic neurons in the brains of schizophrenia patients suggests that a possible strategy to normalize the deficit of RELN and GAD1 expression in schizophrenia is to selectively inhibit DNMT enzymatic activities.
One of the regulatory mechanisms that control gene expression by DNA methylation is the accessibility of transcription factors to their target DNA recognition sites. This accessibility depends on nucleosomal integrity regulated by the acetylation status of histone tails. This, in turn, is governed by a balance of the activities of HATs and HDACs. Studies in the field of cancer therapy suggest that the increased DNMT activity observed in tumor cells can be countered by altering the interaction of DNMT with HDACs. Hence, HDAC inhibitors may prove to normalize possible epigenetic DNA-cytosine methylation defects in schizophrenia.
Valproate is most frequently prescribed as a coadjuvant in the treatment of schizophrenia (71). Because valproate is thought to normalize prefrontal cortex levels of GAD67 and RELN by inhibiting HDACs, one may infer that reduced GABAergic transmission may be one of the putative etiopathogenic mechanisms responsible for the expression of psychotic symptoms in schizophrenia. The beneficial effects in the treatment of schizophrenia obtained with valproate suggest that HDAC inhibitors may represent new opportunities to mitigate vulnerability to schizophrenia among high risk individuals.
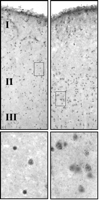
In situ hybridization of DNMT1 in human prefrontal cortex. Signal obtained following in situ hybridization of digoxygenin-labeled DNMT1 antisense oligonucleotides sections obtained from a non-psychiatric subject (left) and a schizophrenia patient (right). Lower panels show magnifications of boxed sections above.
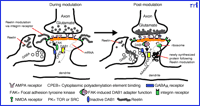
A model for reelin-mediated activation of mRNA translation in pyramidal neuron dendritic spines. Reelin secreted from GABAergic interneurons by a constitutive mechanism interacts with an integrin receptor initiating a cascade that activates translation of spine-resident mRNAs. Reelin is located on postsynaptic densities and initiates a signaling cascade involving the activation of focal adhesion kinase (FAK), phosphorylation of DAB1 and subsequently cytoplasmic polyadenylation element binding protein (CPEB), mRNA polyadenylation and subsequent translation. (Modified from 53.)
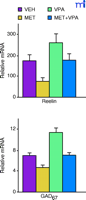
The downregulation of RELN and GAD1 by methionine (MET) in mice is normalized by valproate (VPA).l-Methionine (MET, 6.6 mmol/kg s.c.), valproate (VPA, 2 mmol/kg s.c.) or vehicle (VEH, 0.1 ml/10 g of body weight, s.c.) was administered twice a day for 15 days. RNA was isolated from the frontal cortex and mRNAs were quantified using competitive RT-PCR with internal sequence-specific RNA standards. For details see Tremolizzo et al. (50).
Acknowledgments
This work was supported by grants from the National Institutes of Health MH062188 to EC, MH062188 to AG and MH062682 to DRG.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Alessandro Guidotti, MD, (left) is Professor of Biochemistry in Psychiatry, Dennis R. Grayson, PhD, (center) is Associate Professor of Molecular Biology and Neuroscience, and Erminio Costa, MD, (right) is Scientific Director, Psychiatric Institute, and Professor of Biochemistry in Psychiatry at the University of Illinios at Chicago.

