Reciprocal Interaction of Sleep and Synaptic Plasticity
Abstract
Synaptic plasticity underlying learning and memory has been proposed, on the basis of several experimental approaches, to be intimately related with sleep: 1) The idea that sleep contributes to stabilization of acquired memory arises from numerous studies depriving subjects or animals of sleep. 2) Evidence from developing technologies supports “offline” reprocessing of recent experiences during sleep. 3) Recent analysis of the thalamocortical system establishes the reciprocal observation that sleep itself is a plastic process affected by waking experience. This overview synthesizes these converging perspectives across a variety of brain regions and species. We propose the developing visual pathway as a fruitful model for comprehensive understanding of sleep and synaptic plasticity.

Introduction
Long assumed to be little more than a silent period of rest for the brain, sleep remains something of a biological mystery. Clearly, the functions of sleep are essential to life, as it is evolutionarily conserved from lower organisms to birds and mammals. Sleep rebound occurs after sleep loss, and chronic sleep deprivation (two to three weeks) ultimately causes death in rats. Theories of sleep function acknowledge several multifaceted benefits of sleep: brain thermoregulation, energy conservation, brain detoxification, brain development and plasticity, repair of injury, cognitive function, and restorative function (1–3). The sleeping brain may communicate with the body by secreting certain hormones (e.g., growth hormone and cortisol), but such intersystem communication has been definitively established neither in sleep per se nor in the relationship between sleep and memory.
A number of sleep deprivation studies imply that, subsequent to learning, sleep consolidates acquired short-term memory into a long-term form. Rapid advances in various techniques now allow us to probe the functions of sleep more deeply. Recently, human functional imaging, recording of multiple neurons simultaneously, and genetic or pharmacological manipulation of the brain have converged to support the notion that various sleep stages, in diverse animal species and across different learning tasks, function to reprocess recent memory traces and lead to memory consolidation (1). Nevertheless, the fascinating idea that sleep is a requisite for the stabilization of memory and the restoration of learning performance has been open to debate (4), reflecting the complexity of neuronal learning mechanisms and sleep phenomena, both of which span the whole brain.
In this review, we describe recent examinations of the influence of sleep on memory and synaptic plasticity. We also introduce investigations that indicate a reciprocal relationship between experience-dependent synaptic plasticity and sleep. Finally, we review the issue of neuronal replay/reactivation during sleep after particular experiences, which may further establish a connection between sleep and plasticity. We propose that the developing visual system can serve as a powerful model for studying the direct interaction between sleep and plasticity.
General Features of Sleep
Sleep can be divided into two distinct states, slow-wave sleep (SWS) and rapid eye movement (REM) sleep. State-specific electrical brain activity can be detected by electroencephalography (EEG), electrical recordings generated through a gross electrode placed on the surface of the scalp or the brain. SWS is defined by an EEG pattern of high-amplitude, low-frequency fluctuations, consisting of delta (1–4 Hz), spindle (7–14 Hz), and slow oscillations (0.5–1 Hz) that are generated in the thalamocortical loop by distinct mechanisms (5–6). The electrical behaviors of individual neurons in the thalamic nuclei are described by a variety of ionic conductances with particular voltage dependencies and kinetics and reflect the interplay among cortex, thalamic relay nuclei, and the reticular nucleus of the thalamus (5). Depending on membrane potential, thalamic neurons can exhibit either of two distinct modes of action potential firing. In the hyperpolarized state, thalamocortical neurons intrinsically generate rhythmic burst discharge during SWS. When the cells are tonically depolarized, the firing mode switches to a single-spike pattern, which is dominant during the waking period(7).
The EEG that is generated during SWS is likely to be derived from synchronized activities of a spatially and temporally coherent neuronal ensemble driven by corticothalamic feedback projections. The low-voltage, high-frequency EEG patterns characteristic of REM sleep are similar to those of the waking state but are distinguishable by both a regular field oscillation around 8 Hz (i.e., the theta rhythm) prevalent in the hippocampus as well as the total loss of skeletal muscle tonus. Moreover, ponto-geniculo-occipital waves emerge prominently during REM sleep in the lateral geniculate nucleus (LGN) of the thalamus and in the visual cortex (5).
Neuromodulatory systems with broad projections throughout the brain, such as acetylcholine (ACh), norepinephrine (NE), and serotonin (5-HT) systems, stem from restricted nuclei in the brainstem. All three of these display highest activity during waking and decreased firing rates during SWS. During REM sleep, ACh neurons exhibit activity comparable to the waking state, whereas NE and 5-HT neurons cease firing (8). ACh, NE, and 5-HT enhance membrane excitability of thalamic relay neurons and thereby promote tonic single-spike mode (7).
The ratio of time spent in SWS relative to REM sleep differs according to animal species. Adult humans spend approximately 80% of sleep time in SWS and 20% in REM sleep, alternating several times during a night with a cycle of about 90 minutes; in rodents, the sleep cycle is shorter and more fragmented. Under normal conditions, REM sleep always follows SWS, whereas in narcolepsy, marked by excessive daytime sleepiness, the sleep structure is disturbed, resulting in short latency to REM sleep, REM sleep at sleep onset, and cataplexy. Mutation of either hypocretin (i.e., orexin) or its receptor causes the narcoleptic phenotype in dogs and mice (9). People who are awakened from REM sleep often recall, provided they are prompted immediately upon awakening, bizarre dreams. In contrast, dream content associated with SWS is less vividly described, less emotionally charged, and is in general more coherent than that of REM sleep.
In macroscopic terms, the characteristics of sleep change over the course of development. Slow-wave activity emerges during postnatal life, whereas the neotatal period is dominated by “active sleep,” proposed to be a precursor of REM sleep. In the case of humans, the REM-like sleep occupies 90% of the total amount of sleep at birth, and then declines to mature levels (20%). In addition to the influence of developmental stage, sleep is influenced by circadian rhythms governed by the suprachiasmatic nuclei in the hypothalamus and is regulated in a homeostatic fashion (10), with slow-wave delta activity in the initial part of sleep being quantitatively linked to prior wakefulness. Various hormones appear to regulate sleep and are released according to sleep phase; growth hormone, for example, is secreted during SWS (particularly slow-wave delta activity). SWS is also associated with a decrease in local cerebral blood flow in many brain regions, as revealed by positron emission tomography (PET) (11), whereas protein synthesis is enhanced (12, 13).
Sleep and Memory Consolidation
Sleep-Dependent Learning: A Two-Stage Process
Studies at many levels—from behavioral to molecular—suggest that sleep in humans and other mammals contributes to learning(14). The fear-conditioning paradigm is a powerful animal model, in which robust and persistent learning is measurable following a single trial to establish conditioned responses. Recently, sleep deprivation was found to selectively impair memory consolidation in both contextual (e.g., exposure to a new environment) and cued (i.e., by a conditional stimulus) fear conditioning (15). Both contextual and cued forms of learning are amygdala-dependent, but contextual learning is also hippocampus-dependent. Intriguingly, sleep deprivation 0–5 hours (but not 5–10 hours) after training is detrimental to contextual learning, but not to cued learning. Thus, the relationship between sleep and learning is a function of relative timing, the type of learning involved, as well as of sleep stage. As suggested by human declarative memory and procedural memory tasks (16), a dual-process hypothesis has been favored, according to which SWS and REM sleep act differentially (14).
Stickgold and colleagues (17) have confirmed that some learning [i.e., improvement on a texture discrimination task (TDT)] occurs only after a night of sleep (Figure 1A⇓). This form of latent learning does not occur if sleep is prevented, and sleep-deprived subjects fail to improve on the TDT even after two recovery nights (17). It is noteworthy that performance of TDT is actually improved through a night of sleep, because this improvement (i.e., learning) sharply contrasts with the majority of studies that merely establish a role for sleep in preventing the deterioration of performance (1). Interestingly, TDT improvements correlate well with the amount of early SWS and late REM sleep during the course of a night (Figure 1A⇓) (18, 19). This type of perceptual skill learning, originally developed by Karni and Sagi (20, 21), has unique features (e.g., eye-dependence, retinotopic specificity, and orientation specificity) that suggest that learning occurs at pre-attentive stages of the primary visual pathway (22). These findings again support a two-step process in this task, according to which memory consolidation requires SWS followed by REM sleep (14). Surprisingly, a brief nap (approximately sixty to ninety minutes) containing both SWS and REM sleep can improve performance as does a full night of sleep (23). The notion of sleep-dependent “enhancement” of learning has been extended similarly to motor skills (24, 25).
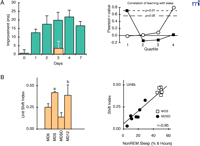
Sleep-dependent improvement of learning and plasticity. A. The improvement of texture discrimination depends on the first night of sleep. (Left): Time course of improvement. Each subject was trained then tested once. Each bar represents a separate group of subjects. Blue bars: control group. Tan bar: subjects deprived of sleep for 30 hours after training and then tested after recovery sleep. (Right): Correlation of learning with SWS and REM sleep across the first night after training. Pearson correlation coefficients between amounts of SWS in each quartile and overnight improvement (filled squares) or for amounts of REM (open circles). Note early SWS and late REM sleep correlate with improved learning. Adapted from Stickgold et al. (17, 18). B. Ocular dominance shift is hampered by sleep deprivation in cats. (Right): Ocular dominance shift of each experimental group. MD6: Kittens assessed immediately after 6 hours monocular deprivation (MD). MDS: 6 hours of MD followed by 6 hours of ad libitum sleep in the dark; MDSD: 6 hours of MD followed by 6 hours of sleep deprivation in the dark; MD12: 12 hours MD. (Left): High correlation between changes in ocular dominance and amounts of SWS in the dark. Adapted from Frank et al. (26).
Another example that links sleep-dependent learning to visual cortical plasticity is found in the well-known ocular dominance assay, which takes advantage of characteristics of the developing mammalian primary visual cortex. In particular, immature animals show a heightened sensitivity to imbalanced visual input during waking. In kittens aged around day P28 (peak of the “critical period”), deprivation of one eye by eyelid suture [i.e., monocular deprivation (MD)] results, within several hours, in a drastic reduction in visual responsiveness of cortical neurons to the closed eye; animals that are visually stimulated (i.e., placed in light) for twelve hours show a significant shift in ocular dominance toward the open eye. Significantly, MD kittens that are visually stimulated for only six hours but are then placed in the dark for another six hours and allowed to sleep show the same shift in ocular dominance; MD kittens that are sleep-deprived during the second six-hour time period, however, lose even the seeds of plasticity from the first six hours (i.e., visual stimulation) as measured by single-unit recording and optical imaging (Figure 1B⇑) (26). The degree of ocular dominance shift positively correlates with the amount of SWS (Figure 1B⇑).
At a cellular level, total sleep deprivation (twelve hours of forced locomotion) reduces long-term potentiation (LTP) of population spikes (evoked by Schaeffer collateral stimulation) in the CA1 region of the rat hippocampus (27). Subsequently, Davis et al. (28) reported a deficit of LTP maintenance upon deprivation of REM sleep. However, the sleep deprivation procedure (immersion from small platform into cold water when muscle tone is lost in REM) itself elevates serum corticosterone levels twofold, and acute stress alone (elevated platform) blocks LTP induction in the CA1 region (29). Thus, interpretation as to the role of sleep should be made carefully.
Nevertheless, hippocampus-dependent learning enhances neurogenesis (30), and toxins directed against proliferating cells can prevent the formation of trace conditioning (31). Intriguingly, cell proliferation, as evidenced by bromodeoxyuridine-positive cells, in the dentate gyrus of adult rat hippocampus is also reduced by sleep deprivation for 96 hours using a treadmill (32). Although stress is known to decrease cell proliferation, serum corticosterone levels do not rise specifically to the treadmill regimen per se. Because exercise can be a stimulator of neurogenesis, an important control is to normalize for total amount of physical activity while still allowing a decrease in neurogenesis to be observable with sleep deprivation (33). Memory impairment brought about by sleep deprivation should thus be treated as a multifaceted biological phenomenon that includes, but is not limited to, LTP.
In the visual system, deprivation of REM sleep extends the period in immature rats during which LTP remains inducible in layer II/III of rat visual cortex (34). This effect is similar to dark-rearing, which prolongs visual plasticity beyond the normal critical period (35) and supports the conclusion that REM sleep, like visual experience itself, promotes brain maturation. However, the selective role of SWS—found to correlate with ocular dominance plasticity in vivo (see above)— has yet to be tested at the cellular level. Moreover, genetic and pharmacological manipulations of LTP and long-term depression (LTD) fail to consistently predict ocular dominance plasticity in kittens and mice (36). It may be difficult to directly apply in vitro synaptic models to in vivo plasticity, as is the case for hippocampus- and amygdala-dependent learning (37).
The REM Sleep Window
Growing experimental evidence supports a preferential role for REM sleep in memory consolidation and learned task performance (38). Increases in the amount of REM sleep are observed after task training in animals that successfully acquire learning but not in unsuccessful or non-learning groups. This increase in REM sleep rates persists for a period of hours to days. REM sleep deprivation after training disturbs memory formation in various types of task. Intriguingly, the deprivation of REM sleep can influence learning and memory only during a restricted period of time, called the “REM sleep window,” after training. The timing of this window varies, ranging over several hours, according to the type of task or training protocol (38). However, Siegel (4) has expressed misgivings about the essential function of REM sleep for memory, suggesting that the effect is due more to stress than to the loss of REM sleep.
Abel and colleagues (39) have suggested possible links among protein synthesis, protein kinase A (PKA), cAMP response element–binding protein (CREB), and the REM sleep window. Specifically, long-term memory for contextual fear conditioning does not develop in mice when protein synthesis is inhibited during a certain time window after training (40). The time course of sensitivity to protein synthesis inhibitors resembles that of the vulnerability of memory to REM sleep deprivation. Similarly, cholinergic blockade experiments fit the idea of a REM sleep window (39). As described earlier, activation of cholinergic neurons occurs during REM sleep as well as in waking. Thus, cholinergic stimulation, PKA activation, and protein synthesis may occur during post-training REM sleep, reflecting common molecular mechanisms for memory consolidation.
REM sleep further draws the attention of sleep researchers due to circumstantial evidence that links REM sleep to synaptic plasticity. LTP readily occurs during peaks of theta oscillations induced pharmacologically in brain slices; LTD is produced at the troughs of the theta rhythm (41). In urethane-anesthetized rats, LTP in the CA1 region is reliably produced on the positive phase of tail-pinch driven theta rhythm, and neither LTP nor LTD occurs at the negative phases or nodes of the theta rhythm (42). In fact, high-frequency stimulation at the negative theta phase can reverse previously elicited LTP. In task-performing rats, the phase of “place cell” firing in the hippocampus established upon exploration of novel environments (i.e., upon learning about “place”) approximates the phase of local theta rhythm observed when the rats later enter REM sleep, whereas cell firing during the exploration of familiar environments (i.e., in the absence of learning) exhibit a reversal of firing phase relative to local theta oscillations in REM sleep (43). Together, these findings seem to corroborate the notion that REM sleep may strengthen synaptic activity related to recent experience while weakening synaptic connections associated with prior experience. The mechanisms of phase reversal remain unknown.
Synchronization of hippocampal and amygdalar theta rhythm occurs in fear-conditioned rats during memory retrieval but not during exploratory behavior (44). Coherent theta-related neuronal communication between nuclei might thus be related to the retention of fear memory. It would be interesting to see if this relationship emerges during REM sleep after learning.
Three distinct classes of hippocampal interneurons discharge, according to behavioral state, with specific phase relationships to the local field potential defined by theta or sharp-wave–associated ripple oscillations (45). This state-dependent firing may reflect information processing and modulation of synaptic connectivity. Interneuron networks within the same class (i.e., GABA-releasing interneurons) coupled to each other via gap junctions can entrain at gamma frequencies typical of waking and function to detect coincident inputs (46). In fact, manipulation of inhibitory networks can trigger the onset of the critical period for ocular dominance plasticity in the primary visual cortex (47). The relevance of the inhibitory system to memory consolidation in sleep is of great interest.
Pontine waves (P-waves), or ponto-geniculate-occipital waves, are generated in the pontine tegmentum and are widely observed in mammals, including humans, primates, cats, and rodents, and precede the onset and continue throughout periods of REM sleep. After avoidance training, P-wave density significantly increases above control levels in rats, and changes in P-wave density positively correlate with improvements in learning (48). When P-wave activation is artificially enhanced by carbachol microinjection into the pontine nuclei after training, rats show improved levels of retention (i.e., higher scores during retest sessions) (49). Therefore, P-wave generation is a possible underlying mechanism for REM sleep–dependent memory consolidation. Local inactivation of the P-wave generator will be required to see whether it is necessary for learning.
Conversely, studies focused on the development of the visual system indicate that REM sleep may protect against excessive plasticity (50). Deprivation of REM sleep in MD kittens amplifies the cell-size disparity normally produced by MD in the LGN: cells in the layer receiving input from the deprived eye become smaller than cells in the layer connected to the open eye. Alternatively, when phasic ponto-geniculate-occipital activities are eliminated from the LGN by bilateral pontomesencephalic lesions (in lieu of REM sleep deprivation), the disparity of cell size across eye-specific laminae is more evident than in sham-operated kittens (51). Accordingly, SWS (26) and REM sleep (50) may respectively affect physiological and morphological plasticity in the primary visual pathway in opposite directions.
Reciprocal Influence of Cortical Plasticity on Sleep
Whereas sleep affects plasticity and memory, new studies indicate that sleep itself is affected by thalamocortical activity in waking cats, mice, humans, and rats (Figure 2⇓). Appropriate development and maintenance of visual cortical receptive field properties requires normal visual experience (52). Normal development of slow-wave activity in SWS depends on visual experience as well (53). Notably, it is sleep quality—not quantity—that is affected here. Raising cats or mice in complete darkness from birth produces a drastic reduction of SWS delta (1–4 Hz) activity in the primary visual cortex without a significant change in other brain regions (Figure 2A⇓). Slightly enhanced somatosensory delta power may in fact reflect increased reliance on tactile information in the dark (see below). The susceptibility of SWS delta activity to sensory deprivation is restricted to the late phase of the critical period of visual cortex assessed by the MD paradigm; placing mature animals (i.e., having aged past the critical period) into the dark for similar periods has little effect on SWS delta intensity. Exposure of cats and mice to a normal lighted condition after dark rearing triggers a delayed ocular-dominance critical period along with gradual increments of delta activity leading to full recovery.
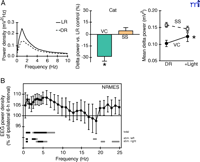
Sensory experience affects quality of subsequent sleep. A. Early sensory deprivation reduces slow-wave activity. (Left) Representative SWS power spectra of light-reared (LR) and dark-reared (DR) cats. DR kittens were raised in complete darkness from birth for 4 months. (Center) Group data. Reduction of delta power after DR is restricted to visual cortex (VC) and not seen in somatosensory area (SS). (Right) Slow-wave plasticity is reversible. Recovery of delta activity after light re-exposure for one to two months. Adapted from Miyamoto et al. (53). B. Sensory stimulation during waking enhances interhemispheric asymmetry of EEG power density. Ordinate indicates EEG power density of hemisphere contralateral to intact whisker side as a percentage of ipsilateral hemisphere (whisker-cut side) during SWS. Lines above the abscissa indicate significant difference between hemispheres. Top line (total) represents statistics for all data, middle and bottom lines for stimulation of intact left or right side whiskers. Adapted from Vyazovski et al. (54).
In contrast to sensory deprivation, sensory stimulation can transiently and modestly enhance SWS activity. Rats that have their whiskers trimmed on one side of the face and are kept awake for six hours in an enriched environment undergo activation of a restricted brain area, namely the somatosensory barrel cortex contralateral to the intact (i.e., untrimmed) side. During subsequent SWS (but not during REM sleep or waking), slow-wave (0.75–6.0 Hz) power increases significantly in the brain region that receives neural input from the untrimmed side of the face (Figure 2B⇑) (54). Similarly, a significant interhemispheric asymmetry of SWS power in the low-frequency range over the sensorimotor cortex shifts to the left hemisphere after a prolonged vibrating sensory stimulus is given to the right hand before sleep onset in humans (55). These studies exemplify a local, experience-dependent facet of SWS activity in response to increased stimulation prior to sleep onset.
Prolonged auditory stimulation results in a widespread increase of spectral power and long-range coherence between fronto-temporal cortical regions during subsequent SWS in humans (56). Also in humans, declarative learning tasks significantly increase the density of sleep spindles relative to non-learning control subjects (57), and asynchronous development of sleep is observed in cortical areas of task-performing monkeys (58). Together, these findings appear to confirm the regional differentiation of SWS activity induced by sensory experience.
The neuronal mechanisms underlying the alteration of sleep activity caused by waking experience remain unknown. Recovery of delta activity in the visual cortex may partially provide a neuronal locus of slow-wave plasticity (53). Because the thalamocortical projection does not remain plastic past the typical critical period even when delayed by dark-rearing (59), intracortical or corticothalamic connections beyond layer 4 may be responsible. Moreover, the level of N-methyl-D-aspartate (NMDA) receptor activation is also implicated by the examination of dark-rearing effects on slow-wave power in gene-targeted mice (53). The influence of cortical plasticity (which is greater in feline than rodent) on slow-wave activity (which exhibited larger changes in kittens than mice) shares a neuronal organization on the same thalamocortical pathway. Sleep is unlikely to be a mere static framework allowing “offline” reprocessing of specific recent events. Sleep itself can be dynamically modulated by experience.
Neuronal Reactivation and Offline Processing During Sleep
Wilson & McNaughton (60) monitored rats running a maze task and made recordings from multiple hippocampal CA1 neurons simultaneously. They found that pairs of cells that fire together when a waking animal occupies a particular place in the environment retain correlated activity patterns during subsequent SWS. Similarly, pairs of neurons with non-overlapping place fields remain uncorrelated during sleep. These observations indicate neuronal reactivation in sleep that is related to experience; neuronal reactivation has also been recorded with respect to temporal sequence, other neocortical structures (61) and different sleep stages (e.g., REM sleep) (62).
Neuronal reactivation associated with sharp waves (and associated ripples), which emerge from the CA3 region, occurs primarily during immobile waking or SWS. The sharp wave/ripple is assumed to induce synaptic modification at target neurons and to transfer stored information about recent waking experience from the hippocampus to the cortex (63). In fact, coactivation of hippocampal ripples and neocortical spindles monitored by local field potentials is observed and is correlated temporally across both sites during SWS in rats and mice (64, 65). Neuronal events seem to be expressed in a time-compressed manner during SWS, and quantified correlations of simultaneously recorded neurons gradually decay over several tens of minutes [reviewed in (66) ].
Studies of song-related neuronal activity in the brain of the zebra finch demonstrate sleep-dependent modulation. During a critical period for song learning, a young bird memorizes his tutor’s song (sensory learning phase) and then rehearses to refine his own song using auditory feedback (sensorimotor learning phase). Likewise, adult male zebra finches require auditory feedback to maintain their song (67). Neurons in two motor control nuclei, the nucleus robustus archistriatalis (RA) and the nucleus of the high vocal center (HVc), display sharp selectivity to an individual bird’s own song in the anesthetized or sleep state, despite the general attenuation of sensory responsiveness. Conversely, auditory responses of the neurons in these nuclei to the bird’s own song are substantially suppressed in the waking state (Figure 3A⇓) (68, 69). Thus, sleep paradoxically promotes selective responsiveness to incoming sensory information, perhaps allowing song learning–related nuclei to better communicate with each other.
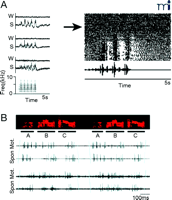
Modulation of auditory activity and song replay during sleep in vocal motor system of songbirds. A. Auditory response is gated by sleep. (Left): Rectified averaged multiunit response to multiple presentations of bird’s own song (BOS) in the nucleus robustus archistriatalis (RA). Recording of 3 sites from one adult bird. W: during waking; S: during sleep. (Right): Raster plot of single RA unit activity. The response to BOS developed as the anesthetic (urethane: arrow) took effect. Adapted from Dave et al. (68). B. Neuronal replay during sleep. Simultaneous recordings from two neurons in RA. Color spectrograph of the song that was sung (Top). Raw traces of neuronal activity during sleep (Spon). Premotor activity during singing (Mot). Adapted from Dave and Margoliash (70).
In addition to the gating function of sleep, neurons in the RA spontaneously generate spike sequences in sleep, quite similar to activity patterns observed during singing (Figure 3B⇑) (70). Because singing-related activities in the RA are tightly coupled to song production, this finding may allow closer examination between sleep replay and song learning. Specific sleep stage was not identified in this study, but it is likely that SWS predominates most of the sleep time in the bird (71).
Molecular evidence has also accumulated that indicates replay during REM sleep. Upregulation of the immediate early gene zif268, for example, is specific to the cerebral cortex and hippocampus during REM sleep of rats that have experienced enriched environments (72). Moreover, induction of LTP in the medial perforant path-dentate gyrus pathway during waking results in the reinduction of zif268 expression during the following cycle of REM sleep in extrahippocampal regions, suggesting the transfer of short-term memory in the hippocampus to a long-term form in the cortex (73). The protein Zif268 is a transcription factor thought to regulate programs of neuronal plasticity; its expression depends upon neuronal activation. Knockout mice that lack the zif268 gene exhibit impairments of late-phase LTP and fail to retain long-term memory (74 ; but see also 75). Human positron-emission tomography also uncovers several brain regions activated during REM sleep, but not SWS, after training of a visuomotor skill (76).
Tononi and Cirelli (77) intensively analyzed the effects of spontaneous and forced waking on the expression of plasticity-related genes using mRNA differential display and cDNA microarray technology. Most known genes are upregulated during wakefulness or sleep deprivation when compared to sleeping animals. Protein expression levels of phosphorylated CREB, activity-regulated cytoskeletal protein, and brain-derived neurotrophic factor, typically associated with synaptic plasticity, are also higher in waking than in sleep. This upregulation during waking depends upon the intact noradrenergic system as well as the immediate early gene products c-Fos and Zif268, and is difficult to reconcile with a sleep memory consolidation hypothesis and the otherwise observed upregulation of zif268 during REM sleep after experience (72). However, it should be noted that expression of plasticity-related genes is not totally lost during sleep, and mRNA levels detected by microarray technology do not necessarily reflect protein levels (78). Moreover, memory consolidation by sleep does not exclude the possibility of selective depotentiation during sleep. Cortical LTD can be induced by low-frequency stimulation reminiscent of the delta frequency during SWS (3, 79).
The ability to induce LTP during given behavioral states has been examined (80, 81; see also 82). These studies suggest synaptic enhancement from perforant path to dentate gyrus is reliably produced during waking (and REM sleep), but is strongly suppressed during SWS. Associations between electrical stimulation of auditory thalamus (i.e., conditioned stimulus) and electrical stimulation of central gray (i.e., unconditioned stimulus) can be formed in the hippocampus during waking or REM sleep, but not SWS (83). Together, these findings demonstrate the potential for replayed activity patterns to induce plastic changes outside of the waking state. Other synapses or patterns of stimulation may be more adequate to observe synaptic plasticity during SWS.
Remaining Issues
Although many studies imply a positive role for sleep in synaptic plasticity, large gaps and critical questions remain (4). Further experiments are needed to better elucidate the relation of sleep (replay) and memory consolidation. As mentioned above, sleep deprivation paradigms face several problems that make the interpretation of experimental results difficult. If sleep is necessary, then deprivation can hardly avoid the nonspecific influences of stress. Time-controlled administration of stress hormones, such as corticosterone (with or without adrenolectomy), should be tested in parallel with learning. New techniques enabling continuous monitoring of the molecular dynamics in the behaving animal (84) may prove to be a useful alternative for observing behavioral modulation of molecular expression without imposing sleep deprivation.
Even if selective REM sleep deprivation is possible, REM sleep pressure may accumulate over the course of deprivation. Latency to REM sleep becomes shorter and the incidence of entry into REM increases, thus finally affecting SWS and distorting sleep architecture. As presented earlier (Figure 1B⇑), each sleep stage may be differentially involved in the memory consolidation process (18), so that disturbances of sleep architecture induced by “selective” REM sleep deprivation may lead to erroneous interpretation. Another pitfall of deprivation studies is the difficulty of differentiating between loss of sleep and gain of prolonged waking. The experimental enhancement of sleep should be complementary to experiments of sleep deprivation, although adequate methods of sleep enhancement are not reliably available. For example, the GABA receptor modulatory agonist diazepam has a well-known sleep-inducing activity, but it also reduces SWS delta activity (85).
Direct evidence linking neuronal replay of experience and memory consolidation is still lacking. Is neuronal replay merely a remnant of neuronal excitation during waking? Part of the difficulty derives from the complex interaction of neuronal circuits responsible for behavioral learning. The formation of hippocampal place cells depends upon integration of sensory information from the environment with the internal brain state (e.g., motivation and memory) (86). Because the exact relationship between place cell activity and spatial memory remains unknown, reactivation of correlated activity within hippocampal neurons does not necessarily mean the replay of memory-related events; replay of activity is observed only after repeated training sessions (62). Similarly, the sequential neuronal activation (i.e., of the sleeping bird) that corresponds to motor-related nuclear activity has been observed in the adult, which is past the critical period for the learning activity (i.e., song). Becuase the adult zebra finch song is not stable after removal of auditory feedback (67), replay may relate more to the maintenance of acquired song, and its role in the learning of a tutor’s song is thus not clear. It would be interesting to see the frequency/intensity of song replay in young, actively learning zebra finches compared with the adult.
Can sleep-stage selective or region-specific perturbation (not simply gross REM sleep deprivation) of neuronal reactivation lead to impairment of learning? For example, electrophysiological experiments reveal that substantial visual- and auditory-evoked responses remain during sleep in the cat visual and rat auditory cortex, respectively (87). Furthermore, the brains of sleeping subjects can be differentially activated by auditory stimulation of different content, as revealed by fMRI (88). The presentation of sensory stimuli may be used to interfere with ongoing information processing during sleep without global changes in sleep architecture. To confirm a role of sleep replay for memory consolidation, it is therefore necessary to test whether giving artificial “neuronal replay” during sleep can mimic learning. Interestingly, conditioning a local increase of fast rhythmic field potentials (20–50 Hz) in somatosensory or visual cortex during waking can also occur during subsequent SWS and REM sleep in cats (89).
Is there any change of functional connectivity, before and after sleep? It will be interesting to explore whether sleep merely stabilizes the connection formed during waking or positively participates in modifying synaptic connectivity through potentiation or depression. The thalamocortical circuit can be a useful model for this purpose. Burst-spiking thalamocortical relay neurons during spindle waves are likely to cause strong depolarization of innervated neurons (79), so that influx of Ca2+ into cortical target cells could stimulate subcellular events, including gene expression and protein synthesis, related to synaptic plasticity. As mentioned, low-frequency activation (around 1 Hz) predominating during SWS appears suitable for the induction of LTD. In addition, synaptic plasticity is controlled not only by mean rate of pre- and post-synaptic firing neurons, but also their relative timing (90). Spike-timing dependent plasticity during sleep should be considered as well.
It still remains unknown how selective neuronal ensembles are activated and what region(s) trigger replay. Replay during SWS is associated with burst activity, which is characteristic of hyperpolarized thalamocortical relay neurons during SWS. It is possible that thalamic activity may drive hippocampal neurons to burst or to gate hippocampal activity into the cortex (64, 91). Thus, examination of the physiological interaction between hippocampus and thalamus would be interesting, in addition to thalamocortical and cortico-hippocampal interactions. Recent studies suggest that the occurrence of thalamic bursting activity is not restricted only to SWS, but is also seen in waking with certain specific behaviors (e.g., whisker twitching) (92), leading to the hypothesis that a descending signal from primary somatosensory cortex triggers thalamic burst activity. This activity deserves to be studied during replay in sleep.
Conclusions
The thalamocortical circuit in the primary visual system offers an attractive system for investigation into the interaction of sleep with plasticity (Figure 4⇓). The visual system is one of the most intensively studied brain structures, both anatomically and physiologically. It exhibits strong experience-dependent circuit reorganization in vivo, which is commonly seen in a variety of mammals, including humans. Thalamocortical circuits process sensory input during waking and spontaneously generate complex patterns of neural activity characteristic of sleep (5). Moreover, at each stage along the pathway (e.g., retina, LGN, and visual cortex), spontaneous synchronized activity is observed in a well-organized fashion (93, 94). Manipulations of visual input can be causally linked to physiological consequences in vivo (e.g., ocular dominance and perceptual learning) more directly than can other learning systems (e.g., hippocampal learning, amygdala fear conditioning, songbird learning, and human procedural learning).
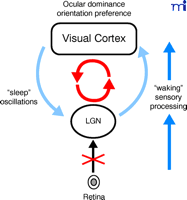
Thalamocortical circuits process sensory input during waking and spontaneously generate patterns of neural activity characteristic of sleep.
Several studies have suggested that neuromodulators are influenced by the vigilance state and affect ocular dominance plasticity (95). Our understanding of the molecular mechanisms of visual cortical plasticity is progressing steadily (96) ; notably, protein synthesis is now known to be required over the several days of MD (97). Recently, genetically manipulated animal models have become available in which the timing of plasticity can be controlled experimentally (47). These models enable us to trace neuronal events in different sleep stages and to test the relationship between sleep and cortical plasticity more directly (98). Hopefully, a profound understanding of the interaction of sleep and plasticity in the visual system will yield basic principles applicable not only to other sensory systems and human perception (20), but also to other higher cognitive function such as verbal memory performance (99).
Acknowledgments
This work was funded by the RIKEN Brain Science Institute, JAPAN.
- © American Society for Pharmacology and Experimental Theraputics 2010
References

Hiroyuki Miyamoto, PhD, (top) is a Staff Scientist in the Laboratory for Neuronal Circuit Development at the RIKEN Brain Science Institute.
Takao K. Hensch, PhD, (bottom) is Group Director of Critical Period Mechanisms Research and Laboratory Head for Neuronal Circuit Development at the RIKEN Brain Science Institute. Send correspondence to TKH. E-mail hensch{at}riken.jp; fax +81-48-467-2306.

