Apolipoprotein AI as Therapy for Atherosclerosis: Does the Future of Preventive Cardiology Include Weekly Injections of the HDL Protein?
A major reason why current guidelines on lipid management focus on reducing the amount of circulating low-density lipoprotein (LDL) cholesterol through the body is because of the availability of powerful LDL-lowering medications, namely the statins, which are ideal agents for long-term use and have proven their effectiveness in large randomized clinical trials (1). However, it is also widely accepted that other aspects of lipid metabolism could produce important therapeutic targets to control the epidemic of atherosclerosis and consequent cardiovascular disease. Interventions on the HDL side of a patient’s lipid profile typically have been based on the concept that raising HDL cholesterol levels will reduce atherosclerosis. This expectation comes from epidemiological observations consistently showing an inverse correlation between HDL cholesterol levels and cardiovascular event rates (i.e., myocardial infarctions) (2, 3). Nonetheless, direct evidence that raising HDL cholesterol levels will reduce cardiovascular risk is scarce and incomplete.
Clinical therapies used to increase HDL cholesterol include smoking cessation, regular physical activity, weight loss, as well as medications such as statins, fibrates, and niacin. The mechanism through which statins raise HDL concentrations is not known and the effect is limited to a few percentage points; however, this “HDL effect” of statins might partly explain the clinical benefits observed in the treatment groups of different clinical trials. For example, in the Air Force/Texas Coronary Artery Prevention Study (AFCAPS/TexCAPS) study, the patients who experienced the greatest benefit from statin usage were individuals with low HDL, and this protective effect might have been due in part to the increase in HDL produced by lovastatin (4). Of the lipid-lowering medications currently used in practice, niacin has the strongest effect on HDL cholesterol (increases average 30–35%). Niacin also exhibits significant salutary effects on cardiovascular disease (5), which can partly be attributed to the effect on HDL levels—although it is well known that this medication beneficially modulates all parameters of the lipid profile.
Fibrates are agonists for the peroxisome proliferator–activated receptor (PPAR) α, a transcriptional regulator of several genes including that for apolipoprotein (apo) AI, the main protein of HDL (6). The use of fibrates effects a 10–20% increase in HDL. Fibrates have also shown evidence of cardiovascular benefits in controlled clinical trials, but these benefits appear to diminish and disappear when baseline LDL levels are higher than 130 mg/dl (7, 8). Among the most exciting new therapeutic molecules on the horizon are cholesteryl ester transfer protein (CETP) inhibitors, agents that can increase HDL by more than 80% (9).
The measurement of HDL cholesterol levels represents only one measure of the status of the reverse cholesterol transport (RCT) system (Box 1).
Reverse cholesterol transport.
Reverse cholesterol transport (RCT) is the pathway through which excess cellular cholesterol is collected from peripheral tissues and transported to the liver via the bloodstream. The carrier of this cholesterol is the high-density lipoprotein (HDL) particle. The HDL starts its maturation process as a lipid-free protein called apolipoprotein AI (apoAI), which is mainly produced by the liver and intestine. ApoAI can engage a membrane transporter on the cell surface called ABCA-1, resulting in the translocation of cholesterol and phospholipids from the membrane to the nascent lipoprotein. An enzyme associated with the HDL, lecithin–cholesterol acyl transferase (LCAT), esterifies the cellular cholesterol and packages it inside the particle, which becomes spherical and progressively larger as the process continues. The large, mature HDL is capable of exchanging cholesterol with very low–density lipoprotein (VLDL) in plasma and, more importantly, can be recognized by the HDL receptor in the liver [the human scavenger receptor class B type 1 (SR-BI)], resulting in the selective removal of the cholesterol cargo and the initiation of a new cycle of maturation for the once again lipid-free apoAI. Even though arterial macrophages do not produce apoAI, they need it to get rid of their excess cholesterol via ABCA-1. Interventions aimed at providing apoAI to the macrophages within the plaque have demonstrated that the vascular effect of modulating RCT does not necessarily require increasing the level of plasma HDL cholesterol.
Therefore, it is possible that, unlike what we have learned with the LDL pathway, where improved metabolism means lower plasma concentrations, interventions that exert large beneficial effects on RCT might only translate into relatively minor increases in plasma HDL cholesterol levels. Taking this idea to its extreme, one could also envision improvements in RCT without any measurable changes in HDL cholesterol levels or, paradoxically, with reductions in HDL (Figure 1⇓). Surprisingly, this has been the case with the apoAIMilano mutation, which differs from wild-type apoAI by only one amino acid: R173C (10). Carriers of this HDL mutant have been identified on the basis of low HDL cholesterol levels and concomitant longevity (11). Human HDL that contains apoAIMilano appears to undergo faster catabolism, which explains the lower levels of HDL cholesterol and apoAI in the plasma of carriers (12). Reportedly, apoAIMilano has a superior ability over normal apoAI to drive cellular cholesterol efflux, the first step of RCT (13). Thus, at the time when almost every clinician thinks of an intervention on the RCT as being necessarily based on raising HDL cholesterol levels, the possibility exists of HDL-based interventions that produce cardiovascular benefits without changing, or even by inducing decreases in, HDL cholesterol levels.
The use of injected apolipoproteins to affect atherosclerosis has been practiced in experimental systems for many years. Injections of apoE, an apolipoprotein similar in structure to apoAI, have a significant effect on reducing the size of lipid–cholesterol plaques in hypercholesterolemic rabbits (14, 15). Similarly, injections of normal apoAI have been said to demonstrate the feasibility of an atherosclerosis-reducing intervention aimed at modulating RCT (16). Also, studies in transgenic mice have revealed that overexpression of human apoAI has a protective effect against diet-induced atherosclerosis (17, 18), and positive results have been observed in studies of adenoviral-mediated overexpression of human apoAI in mice as well (19). In all these cases however, the injection or expression of apolipoproteins had measurable and beneficial effects on plasma lipid concentrations, and this could have easily explained the vascular changes. Injected apoAIMilano has been used in animals to show a direct vascular effect, but because these interventions were undertaken with large amounts of apolipoprotein, they caused increases in HDL cholesterol and total apoAI levels (20, 21). However, the concept that apoAI might influence lipid–cholesterol plaque size and composition irrespective of plasma HDL concentrations has been validated in other studies that showed that the expression of normal human apoAI by arterial macrophages, due to transgenic expression or retroviral transduction of bone marrow cells, significantly reduces atherosclerosis without affecting HDL cholesterol levels (22–24). Therefore, if macrophages acquire the ability to secrete the HDL protein even in small amounts, large changes on the atheroma will follow without increases in HDL cholesterol.
Another problem that has marred the investigations on apoAIMilano is that none of the studies compares the effects of the mutant with that of the normal apoAI, which is in itself capable of activating cholesterol efflux and of stimulating RCT. In this context it is important to emphasize that increasing the plasma level of apoAI is the benchmark of current pharmacological options affecting HDL.
Results of a study by Nissen et al. on the effect of apoAIMilano injections in patients with atherosclerosis have recently been published (25). In this study a small group of patients received either saline infusion or five weekly injections of two different dosages of apoAIMilano-containing phospholipid particles. In just five weeks, two repeat intravascular ultrasound (IVUS) evaluations demonstrated an effect on the plaque reported as a 4% regression in total atheroma (i.e., an accumulated lipid–cholesterol plaque) volume. This study is, in many ways, remarkable, as it shows the clinical applicability of a therapeutic concept that has been tossed around by experimentalists for nearly twenty years, and demonstrates that the arterial plaque in human subjects is a plastic tissue that can undergo quick volume regression. However, the study has limitations in scope and relevance that need to be considered. First, the authors did not feel compelled to evaluate the effect of apoAIMilano vis-à-vis that of normal apoAI. Similar to previous work in experimental animals, these results cannot be interpreted as sure evidence that it was indeed the peculiar structure of apoAIMilano, and not the general apoAI effect, that produced the vascular changes. What is even more surprising is that the authors did not monitor changes in HDL cholesterol or any other plasma lipid parameters. This is a tremendous missed opportunity, because correlating the vascular effects with any changes in plasma HDL cholesterol levels would have allowed for a better prediction of the future therapeutic use of this medication. For example, if the injected apoAIMilano raised HDL, this would support the widely held notion that HDL increases are good, but at the same time, might make the results less physiologically relevant considering that apoAIMilano depresses circulating HDL levels in carriers by virtue of its quick turnover. On the other hand, if the HDL cholesterol levels were reduced by the injections of apoAIMilano, this intervention might not receive widespread acceptance in clinical practice, as physicians may not be ready to adopt a strategy that can only be monitored through a change in a biochemical parameter (HDL) that would appear to move in the wrong direction.
Another interesting aspect of this study was the absence of a dose-dependent effect of apoAIMilano on the IVUS outcomes. The dosages used were hefty, either 15 or 45 mg/kg, and would be expected to increase significantly the concentration of total apoAI in plasma. However, because the effect of normal apoAI on atherogenesis (i.e., plaque formation) is dose-dependent, these results indicate either that apoAIMilano has a dominant-negative effect on the plasma apoAI pool, or that even the smaller injection dose of apoAIMilano was above the pharmacologic titration range. It is impossible at this time to know whether the protective effects of the mutant protein were indeed due to its specific impact on RCT or to a non-specific increase in the apoAI pool.
Finally, the importance of this study is somewhat reduced by the small number of patients enrolled, which resulted in the lack of statistical significance for the comparisons between treatment and placebo groups. However, the effect of the active intervention on atheroma volume in individual patients (in comparison to their own baseline levels) was undeniably positive and significant, whereas standard angiography on these same patients failed to show any differences due to treatment. This confirms that IVUS is an excellent methodology to investigate coronary status (26), with more sensitivity than angiography and possibly better predictive power for clinical outcomes. This latter point remains to be proven, however, as small volume changes in early plaques might not translate into significant clinical benefits.
The best stance for the informed clinicians and clinical researchers eager to learn about new therapeutic interventions in atherosclerosis is to wait for better and more complete data on the usefulness of injectable apoAI (either the wild-type protein or its natural or synthetic variants) in improving RCT and vascular health. To provide the final evidence of benefits, a study should be designed with clinical outcomes as primary endpoints. Given the rapidity of the anatomical changes induced by apoAIMilano, effects on endpoints such as angina could be detected in less than a year using the right population size. Such a study would address the untapped potential in practice for an acute and aggressive pharmacologic modulation of the arterial plaque.
For cardiovascular interventions based on apoAI injections to become standard practice in preventive cardiology, we must accept that the goal of these therapies is to activate cholesterol efflux from the plaque. We should be prepared to discover that, in some instances, monitoring changes in plasma lipoprotein levels induced by pharmacological interventions may not be an adequate way to assess clinical benefits. What is needed to resolve the issue of potential dissociation between HDL/apoAI levels and clinical effects is a reliable methodology to measure RCT.
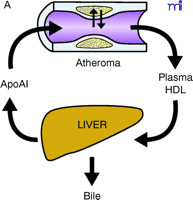
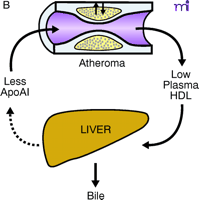
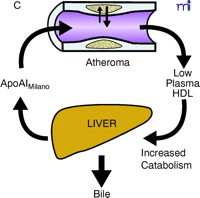
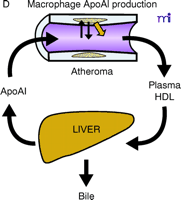
ApoAI, HDL cholesterol, and the atheroma. A. Under physiological conditions, the production rate of apoAI is the main determinant of HDL cholesterol levels, and both correlate well with the extent of cholesterol efflux from peripheral cells. The macrophage cluster in the atheroma is depicted as an example. B. The most common reason for low HDL is reduced production or increased elimination of lipid poor apoAI, resulting in impaired cholesterol efflux from peripheral cells. C. The apoAIMilano reduces HDL through increased catabolism of mature HDL. This is not associated with vascular deterioration because the production rate of apoAI is normal and its ability to collect cellular cholesterol is increased. D. A strategy based on increasing the amount and availability of normal apoAI in the vessel wall results in vascular benefits without any changes in plasma HDL cholesterol levels (see text for details).
Acknowledgments
The authors are supported by National Institutes of Health grants HL53989, HL58427, HL57986, and HL65405. MFL is an Established Investigator of the American Heart Association. We would like to thank Dwayne Dove for helpful discussions and for the production of the figure.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

MacRae F. Linton, MD, (right) is a graduate of the University of Tennessee, Memphis. He completed his Residency in Internal Medicine at Vanderbilt University and his Fellowship in Endocrinology at the University of California, San Francisco. In 1993 he joined the faculty of the School of Medicine at Vanderbilt University as an Assistant Professor in the Division of Endocrinology and Diabetes and in November of 1999 became the co-Director of Preventive Services including the Atherosclerosis Research Unit and the Heart Disease Prevention Program. Dr. Linton co-founded the Vanderbilt Lipid Clinic and is the Director of the Vanderbilt Lipid Clinic. Email: macrae.linton{at}vanderbilt.edu; fax (615) 936-3486. SF and MFL run their clinical and research enterprise in partnership. Their clinical interests include the management of dyslipidemic patients, and involvement in determining new mutations that cause altered lipid levels in humans. Their research focuses on the pathogenesis of genetic dyslipidemias, the early cellular events of atherogenesis, and gene therapy approaches to atherosclerosis.

Sergio Fazio, MD, PhD, (left) is a graduate of the medical school of the University of Rome, Italy. He completed his Residency in Internal Medicine and a Fellowship in Metabolic Diseases at the same institution. In 1985, he undertook a graduate study program in Molecular Biology at the University of Siena, Italy, and completed it at the University of California, San Francisco (UCSF). In 1993, he joined the faculty of the School of Medicine at Vanderbilt University as an Assistant Professor in the Division of Endocrinology and Diabetes, and in November of 1999 he became the co-Director of Preventive Services including the Atherosclerosis Research Unit and the Heart Disease Prevention Program. He co-founded the Vanderbilt Lipid Clinic and is the Director of the Vanderbilt Lipid Laboratory. Email: sergio.fazio{at}vanderbilt.edu; fax (615) 936-3486.

