A Neurogenic Theory of Depression Gains Momentum
No neuropathological hallmarks exist for depression, despite decades of research investigating the cause of this significant public health problem. The hippocampus increasingly has been implicated as a key brain system involved in depressive disorders, as it influences many functions impaired in depressed individuals, including mood and cognitive difficulties, adverse responses to stress, and altered neuroendocrine functions. An emerging hypothesis linking hippocampal neurogenesis and its control to the cause and subsequent cure of depression is attractive to neuroscientists and clinicians alike. Alterations in hippocampal morphology––including decreased synaptic plasticity and decreased neurogenesis––occur both in animal models of depression and in humans (1). The hippocampus is one of only two areas in the brain clearly shown to exhibit neurogenesis throughout the life of mammals, including humans (2, 3). Recent evidence demonstrates that these newly generated neurons display action potentials, establish synapses, and may function as mature dentate granule cells (4).
The functional purpose of continued neuronal birth has received much speculation. As environmental enrichment increases the rate of neurogenesis, one possibility is that the decreased hippocampal neurogenesis found with depression may reflect an impaired ability to cope with novelty and complexity (5). This hypothesis is supported by work correlating neurogenesis with reactivity to novelty where hyperactive rats exhibited lower rates of cell proliferation in response to novelty compared to low-reactive animals (6). This linkage between neurogenesis and depression was recently advanced by a report from the laboratory of Rene Hen and colleagues advocating a causal relationship between increased neurogenesis and amelioration of depressive behavior in mice (7).
Neurogenesis is a complex series of events resulting in the generation of new neurons from a pool of more primitive progenitor or stem cells (Figure 1⇓). This process occurs during development of the nervous system and, to a more limited extent, during adulthood (8). The rate of adult neurogenesis is not static, but changes in response to environmental factors. Whether such modulation affects the rate of proliferation or the survival of new neurons will differentially influence the net outcome on neuronal number (9). It is the integration of these differentiated, new neurons into the existing neural circuitry that will likely determine the long-term effect of neurogenesis. A variety of stimuli may positively or negatively alter neurogenesis (Figure 2⇓) (10–14); for example, stress is a potent reducer of adult neurogenesis (15–17). Given that stress is a factor implicated in psychiatric illness such as depression, it is plausible that this stress-induced reduction in neurogenesis may be one aspect of the pathophysiology of depression.
Many antidepressants stimulate hippocampal cell proliferation, the first stage of neurogenesis (18, 19). Indeed, the latency of onset of clinical efficacy with antidepressant medication may be the time required to restore “depressed” neurogenesis and allow for maturation of new neurons. Different classes of psychotropic drugs facilitate hippocampal neurogenesis through modulation of various neurotransmitters suggesting that a common molecular mechanism may underlie different antidepressant intervention strategies (Table 1⇓). Some of the possible mechanisms under intense scrutiny include signaling pathways such as the cAMP-dependent protein kinase (PKA)–cAMP response element binding protein (CREB) cascade and the regulation of effectors downstream of growth and neurotrophic factors (20–24). Thus, alteration of neurogenesis in response to different drug administration may provide insight into the mechanisms regulating neurogenesis.
Although the role of neurogenesis in human depression is of primary interest, for ethical reasons it is not possible to design studies using human subjects because our current method of detecting new neurons requires administration of thymidine analogs. Therefore, it is essential to utilize animal models to elucidate those adaptive molecular responses that lead to proliferation and neurogenesis and appear to ameliorate depressive behavior. Animal models of depression, though numerous, are elusive in their ability to define depression and instead rely on various behavioral outcome measures. Most animal models of depression quantify the behavioral effects to various stressors. In fact, antidepressant efficacy is deduced from the medication’s ability to diminish such behavioral effects. These behaviors closely resemble human symptoms of depression and animals exposed to natural and laboratory stress exhibit diminished appetite, sexual drive, grooming, and social behavior (25).
Santarelli et al. adapted a novelty-suppressed feeding test as their measure of the behavioral effects of antidepressants (7). In this test, latency-to-begin-eating is used as an index of anxiety-like behavior. Mice receiving antidepressants exhibited a 35% reduction in latency compared to those receiving vehicle. This reduced latency was correlated with a 60% increase in proliferating hippocampal cells. The specificity of fluoxetine to reduce latency was shown by their ineffectiveness on 5-HT1A receptor-null mice. To demonstrate the requirement of neurogenesis for this effect, the authors blocked hippocampal neurogenesis by localized X-irradiation, which abolished the effect of antidepressants on latency.
Because blocking neurogenesis blocked the behavioral effects of antidepressants, this study provides evidence for a causal effect for the rate of neurogenesis on depression (7). However, there are some limitations to the strength of this interpretation, primarily because the mice in the report of Santarelli et al. were not exposed to stress and were thus baseline, “non-depressed” animals. Some groups of mice were subjected to stress to evaluate the integrity of their limbic structures following X-irradiation, but no data for the effects of antidepressant administration was reported for these groups [online supplementary material to (7)]. For instance, it is interesting that Santarelli et al. found that both proliferation and differentiation of new neurons was increased with antidepressants [online supplementary material to (7)], in contrast to Malberg and Duman (9) who found that stressed animals showed elevated proliferation but not differentiation or survival of new neurons in response to antidepressant administration. Differences in the animal models of depression studied may account for these observed differences in neuronal differentiation and underscore the need for additional lines of evidence to be presented to strengthen the proposed relationship between neurogenesis and depression.
If diminished hippocampal neurogenesis is indeed the neuropathological hallmark of depression, then antidepressant administration may be only one way to treat this disease. It may be possible that other conditions that increase neurogenesis (Figure 2⇓) could likewise provide antidepressant effects. Additionally, if depression is a disease of neurogenesis, then it may be possible to predict risk factors for depression based on factors that decrease neurogenesis (Figure 2⇓). The neural circuitry involved in depression is widespread and it may be too soon to conclude that hippocampal neurogenesis is exclusively implicated as the neurobiological basis of depression. Although the assertion of Santarelli et al. linking depression and neurogenesis is provocative and provides a conceptual framework for further investigation, it may be premature to define such a causal relationship. The development of more effective pharmacological intervention for depression aimed at modulating neurogenesis will depend on further elucidation of the mechanisms regulating neurogenesis and the determination of the functional role of newly generated neurons in the adult brain.
Response of Neurogenesis to Administration of Neuroleptic Drugs1
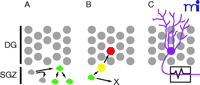
Process of neurogenesis. Neurogenesis in the adult hippocampus is a process that includes progression from cell proliferation through lineage commitment and functional integration. A. Self-renewal and Proliferation Phase. Resident neural stem cells in the subgranular zone (SGZ) below the hippocampal dentate gyrus (DG) occasionally undergo cell cycle, producing progeny that either retain their stem cell-like nature (self-renewal) or begin a process of lineage commitment as neural progenitor cells (green). The progenitor cells act as a reserve of cells that can be expanded under the influence of environment factors, such as antidepressants. B. Survival and Differentiation Phase. Under appropriate environmental conditions, some progenitor cells (green) progress in their lineage commitment (yellow) and move into the granule cell layer where they begin to express markers characteristic of mature neurons (red). This represents the process of differentiation into neurons. Other progenitor cells may commit to become astrocytes or die (X). This process is typically complete within one or two weeks from the progenitor cell’s “birthdate” when it exits from cell cycle. C. Maturation and Functional Integration Phase. Newly generated neurons eventually establish a morphology consistent with neuronal function such as the elaboration of a dendritic arbor, formation of dendritic spines and synapses, axonal extension, and the ability to conduct action potentials.

Some environmental factors reported to modulate the rate of neurogenesis. See (10) for a complete review.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Rosanne Thomas, MSPT, (left) is an Assistant Professor of Physical Therapy at the Chicago Medical School, teaching neurological treatment approaches in the doctor of physical therapy program. She received an advanced masters degree in neurological physical therapy in 1996 and maintains a part-time clinical practice focusing on improving functional outcomes for patients with neurological dysfunction. In addition to her academic and clinical duties, RT is a PhD candidate in the Neuroscience Graduate Program at the Chicago Medical School and is doing her dissertation work in Dr. Peterson’s lab. Her work focuses upon the environmental modification of adult neurogenesis in models of acute stress. Given her diverse professional background, RT’s goal is to bridge the gap between basic science research and clinical application.
Daniel A. Peterson, PhD, (right) is an Associate Professor of Neuroscience at the Chicago Medical School where he is director of the Laboratory for Neural Repair and Neurogenesis and Director of the University’s Microscopy and Imaging Facility. DP earned his PhD at the University of Otago School of Medicine (New Zealand) and completed a post-doctoral fellowship in the Department of Neurosciences at the University of California, San Diego. After serving as a Staff Scientist in the Laboratory of Genetics at the Salk Institute for Biological Studies in La Jolla, California, DP joined the faculty of the Chicago Medical School in 1998. His research focuses on the regulation of neurogenesis in the adult brain, particularly as a function of aging, and the development of strategies to repair the nervous system using a combination of stem cell and gene therapy approaches. DP’s research is supported by grants from the National Institute on Aging. Address correspondence to DAP. Email: Daniel.Peterson{at}finchcms.edu; fax (847)-578-8545.

