Sodium-Dependent Neurotransmitter TRANSPORTERS: OLIGOMERIZATION as a Determinant of Transporter Function and Trafficking
Abstract
Constitutive oligomer formation appears to be the rule for the neurotransmitter:sodium symporter (NSS) family of proteins. The propensity to form oligomers is a prerequisite for NSS proteins to pass the rigid mechanisms of quality control in the endoplasmic reticulum. Moreover, recent findings suggest that correct trafficking to the plasma membrane appears to rely on the interaction of NSS homo-oligomers with components of the COPII-vesicle machinery. The transporters present at the plasma membrane are most likely organized in a tetrameric arrangement, as a dimer of dimers. In this review, we will address ongoing efforts to unravel the underlying mechanisms of oligomer formation at the molecular and cellular levels, and we will discuss oligomerization in terms of transporter function.

Introduction
The action of released neurotransmitters is terminated by a group of highly specialized membrane proteins, the neurotransmitter transporters. The sodium-dependent neurotransmitter transporter subfamily, or neurotransmitter:sodium symporter subfamily (i.e., NSS 2.A.22, according to a recently published nomenclature that will be used herein; 1), comprises transporters of monoamines (i.e. dopamine, norepinephrine, and serotonin) and of amino acid neurotransmitters [(e.g., γ -aminobutyric acid (GABA)], as well as transporters of osmolytes and other uncharacterized substrates (2). The NSS proteins typically share a common structural motif consisting of twelve transmembrane helices and act to terminate neurotransmission by transporting synaptic substrates back into the releasing presynaptic cell, effectively coupling neurotransmitter reuptake to the sodium gradient across the plasma membrane (3).
Since the seminal discovery of the inhibitory effects of diverse psychotropic and therapeutic drugs on norepinephrine uptake, by Axelrod, Whitby, and Hertting (4), the monoamine transporters have gained great clinical relevance as the targets of antidepressants (5). The rewarding properties of psychostimulants such as amphetamine, methamphetamine, and congeners such as methylenedioxy-methamphetamine (better known as “ecstasy”; see 6) result from the ability of these monoamine substrates to induce efflux of dopamine and serotonin, in a process of reverse transport, by their respective transporters (7, 8). In contrast, the psychostimulant cocaine exerts its effects by blocking the reuptake of dopamine and other monoamines. Reuptake of GABA, the principal inhibitory neurotransmitter in the brain, is blocked by the antiepileptic drug tiagabine (5).
Homomultimerization of NSS-Family Proteins
Increasing evidence suggests that NSS-family members are organized into oligomeric complexes (9, 10). Previously, radiation inactivation (11, 12) or detergent solubilization and subsequent separation by chromatography (13) yielded the first clues to the complex nature of the NSS quaternary structure (reviewed in 14). More recent studies using coimmunoprecipitation have also supported an oligomeric structure for the serotonin and dopamine transporters (15, 16). In addition, the rat serotonin transporter was crosslinked into oligomeric species (17), although the specific residues that were crosslinked could not be identified.
We have shown that the human dopamine transporter (DAT) can be oxidatively crosslinked into homodimers that retain transport functionality (18). Because Cys306, just N-terminal (i.e., extracellular) to the sixth transmembrane segment (TM6), is both necessary and sufficient for crosslinking, this TM6 oligomeric interface must be symmetrical. The existence of a canonical dimerization motif in TM6 of DAT and many related transporters supports this conclusion (see below). In addition, we have reported that a zinc binding site engineered at the extracellular end of TM6 results in potent zinc inhibition of dopamine uptake (19), which may reflect a requirement for a conformational change at the dimeric interface for transport activity. More recently, we have used cysteine crosslinking strategies to identify additional oligomeric interfaces and to further define the quaternary structure of DAT. In addition, studies using dominant negative forms of DAT, as well as of the serotonin transporter (SERT) and norepinephrine transporter (NET), have also supported the existence of transporter oligomers (20–22).
Oligomer Formation in Living Cells
Fluorescence resonance energy transfer (FRET), first described by Förster in 1948 (23), has recently been applied to the study of protein–protein interactions in living cells (Figure 1⇓) (24). FRET is a quantum physical phenomenon that occurs if two fluorophores are in sufficiently close proximity (<100 Å) and in an appropriate relative orientation, so that one excited fluorophore (i.e., the donor) is able to transfer its energy to a second, longer-wavelength fluorophore (i.e., the acceptor) in a non-radiative manner (24, 25). The green fluorescent protein (GFP), originally isolated from Aequoria victoria (26), and its spectral variants the yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP), have been fused to a wide variety of proteins, which remain functional. Moreover, the use of CFP and YFP together can enable investigations of physical association between proteins by FRET; several membrane proteins, including G protein–coupled receptors (27) and ion channels (28), have been successfully examined using FRET-based approaches. We have used CFP and YFP to examine the oligomerization of NSS-family members (Figure 1⇓) with three different FRET-based approaches, including ratio imaging, donor photobleaching, and donor recovery after acceptor photobleaching (25). FRET has established the existence of oligomers in the case of the human SERT and the rat GABA transporter-(GAT)-1 (29), and more recently, the human DAT (16). These studies show that SERT, GAT-1, and DAT normally exist as oligomers not only in the plasma membrane of living cells, but also through the course of biosynthesis and protein trafficking.
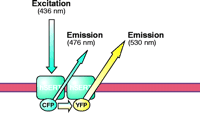
FRET methodology. Fluorescence resonance energy transfer (FRET) is a quantum physical phenomenon that occurs only if two fluorophores are in sufficiently close proximity (<100 Å) and in an appropriate relative orientation. When these two criteria are met, an excited fluorophore (donor) can transfer its energy to a second, longer-wavelength fluorophore (acceptor) in a non-radiative manner. Among the spectral variants of the green fluorescent protein (GFP), the cyan and yellow fluorescent proteins (CFP and YFP) enjoy widespread use. The figure shows these fluorescent proteins fused to the N terminus of SERT. Enhancement of the acceptor (YFP) emission results from transfer of energy from the donor molecule (CFP); the donor emission diminishes concomitantly. In this case, the energy transfer is indicative of physical association of two (or more) SERT molecules in the plasma membrane.
Structural Bases of Oligomerization and Oligomer Stoichiometry
The GxxxG Motif in TM6
We have identified in the intracellular half of TM6 of DAT a well characterized dimerization motif, G(V/I)XXG(V/I)XX(A/T), originally identified in glycophorin A (30–32). This dimerization motif is found not only in DAT, but in other neurotransmitter transporters as well (Figure 2⇓). The second glycine of the motif (i.e., Gly327 in the DAT sequence) is conserved in all NSS proteins and is the only residue in the glycophorin A motif that is indispensable to dimerization (30). The existence of this motif in TM6 of DAT, the extracellular end of which is susceptible to disulfide crosslinking, suggests to us that this motif functions in DAT as it does in glycophorin A, namely, to promote a TM6–TM6 interaction essential to multimerization. Mutation of the second glycine to valine (i.e., G327V) completely prevents expression of DAT at the cell surface (18). Mutation of the first glycine to valine (i.e., G323V), moreover, greatly decreases surface expression, and the small amount of the G323V that reaches the cell surface, although susceptible to disulfide crosslinking, is nevertheless completely nonfunctional as a transporter (18). The prevention of cell surface expression by interfering with oligomerization has been observed for other oligomeric proteins (33), and so the functional dimerization motif of TM6 may well be necessary for efficient trafficking of DAT to the cell surface (see below).
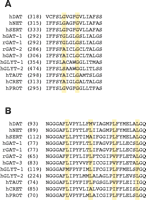
Sequence alignments of transmembrane domains 6 and 2. The sequences of NSS-family members are shown for transmembrane domain 6 (Panel A) and transmembrane domain 2 (Panel B) and indicate the relative conservation of the glycophorin-like dimerization motif (GxxxG, Panel A) and the leucine heptad repeat (LxxxxxxL, Panel B). Note that the first glycine of the GxxxG-motif is substituted by an alanine in some sequences (30). The amino acid position of the first residue of each sequence shown is given in parentheses. (DAT, human dopamine transporter; NET, norepinephrine transporter; SERT, serotonin transporter; GAT, GABA transporter; GLYT, glycine transporter; TAUT, taurine transporter; CRET, creatine transporter; PROT, proline transporter; h, human; r, rat).
That the GxxxG motif is highly conserved in other neurotransmitter transporters suggests that a similar symmetrical TM6–TM6 interface may support the oligomerization of other proteins, but this commonality has not yet been explored experimentally. In glycine transporter-(GLYT)-2 and several other neurotransmitter proteins, the first glycine of the dimerization motif is not conserved, but rather is represented by a serine or alanine residue (Figure 2⇑). Alanine is indeed tolerated at this position in glycophorin A, but substitution with serine has not been evaluated. Curiously, GLYT-1 and GLYT-2 have been inferred to be monomers at the cellular membrane (34, 35), despite the presence of the GxxxG motif in GLYT-1. Additional studies by FRET and crosslinking approaches to evaluate the oligomeric status of glycine transporters are thus warranted.
Transmembrane Domain 4 of DAT
Recently, we have demonstrated the presence of a second symmetrical interface in DAT, in this instance involving TM4 (36). Copper-induced cysteine crosslinking of wild-type DAT in intact cells results in electrophoretic bands indicative of dimeric, trimeric, and tetrameric species. Cysteine-depleted variants of DAT, retaining either Cys243 (i.e., of TM4) or Cys306 (i.e., of TM6) alone, were prepared by site-directed mutagenesis and then subjected to conditions that promote intermolecular disulfide bond formation; in each case, dimerization occurred. When both Cys243 and Cys306 were preserved in the otherwise cysteine-depleted DAT, patterns of crosslinking were indistinguishable from those of the wild-type protein, indicating the existence of dimers, trimers, and tetramers. These results collectively indicate both TM4–TM4 and TM6–TM6 interfaces between DAT molecules, so that DAT likely exists in the plasma membrane as a dimer of dimers (Figure 3⇓). The high-affinity cocaine analog MFZ 2–12 and other DAT inhibitors, including benztropine and mazindol, prevent intermolecular crosslinking at Cys243 (i.e., TM4), whereas they have no significant effect on oxidative crosslinking at Cys306 (i.e., TM6). The impairment of crosslinking effected by the inhibitors, although conceivably due to dissociation of the DAT tetramer, more likely results from a conformational change at the TM4 interface. This conclusion is based on preliminary crosslinking and protection data on an extensive series of TM4 cysteine substitution mutants (H. Hastrup and J. Javitch, unpublished results).
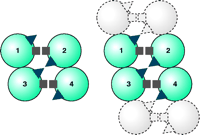
Schematic representation of DAT as a dimer of dimers. Each circle represents a DAT molecule, with four molecules in a homotetramer numbered 1–4. The squares represent one symmetrical interface (i.e., Cys306). The triangles represent a second interface (i.e., Cys243). Note in the left that the triangles in molecules 1 and 4 do not exist at a symmetrical interface, whereas the triangles in molecules 2 and 3 do so. Squares in all molecules can crosslink. To the right, another set of dimers is shown shaded in gray as they might exist in an array, and in this case both symmetrical interfaces can be repeated within in the array. Reproduced from (36).
Given the existence of two distinct symmetrical interfaces (i.e., TM4–TM4 and TM6–TM6 disulfide bridges), the minimal level of quaternary structure established by DAT in the plasma membrane must be a dimer of dimers (Figure 3⇑). Although there is no experimental evidence that DAT exists as more than a tetramer, it is unclear geometrically what would limit assemblies of DAT to the level of tetrameric association so as to prevent more extended arrays. Indeed, given the existence of four Cys243 residues among the four promoters in a tetrameric association, two of which can be crosslinked symmetrically, there must be two other Cys243 residues per tetramer that are not in sufficient proximity to each other to crosslink and therefore do not appear to participate in higher-order oligomerization (9) (Figure 3⇑). Both the N and C termini of DAT and related transporters interact with other proteins (37, 38), and these interactions may prevent DAT from assembling into more extended arrays. It is also conceivable that DAT forms higher-order arrays that go undetected for technical reasons.
Leucine Heptad Repreats in TM2
Four tandem repeats of a leucine heptad (Lxxxxxx; i.e., four leucines, each separated by six amino acids) is a structural motif that underlies oligomerization in phospholamban (39). Originally described in soluble transcription factors (40), this leucine heptad repeat supports the formation of a “leucine zipper.” A perfect leucine heptad repeat is found in the TM2 of the rat GAT-1 (Figure 2⇑) (41). Monoamine transporters including DAT, NET, and SERT contain incomplete leucine heptad repeats in which one leucine is replaced by another amino acid (Figure 2⇑). Several studies support a significant role for leucine heptad repeats in oligomer formation of transmembrane domain proteins (41–47). Thus, by analogy with soluble proteins, a leucine zipper–like structure may play a role in transmembrane protein multimerization.
In the rat GAT-1, we progressively exchanged leucines with alanines (41), and the resulting mutant proteins (i.e., L97A, L83A/L90A, L83A/L90A/L97A and L83A/L90A/L97A/L104A) were tagged with fluorescent proteins. Upon expression in HEK-293 cells, the mutant proteins accumulated in the cytoplasm, and FRET microscopy indicated that they failed to undergo normal intermolecular association (41). Hence, we infer that an intact leucine heptad repeat supports oligomerization of GAT-1, and a similar conclusion has been drawn for DAT (21).
Several objections can be raised, however, regarding the proposal of a leucine zipper–like structural arrangement in the transmembrane domain. First, polyleucine segments show a limited propensity to associate into complexes within lipid bilayers (46). In addition, leucine heptad repeats have been examined in ion channels, such as the shaker K+ channel and L-type calcium channels (48, 49), where leucine heptad repeats were implicated in gating rather than oligomer formation. Thus, the precise physical interactions that involve TM2, and the role that they subserve, remain to be established.
Interactions Near the C Terminus of SERT
Constructs of human SERT that express either the N- or C-terminal half of the protein and a CFP or YFP tag are retained in the cytoplasm (22). Neither construct is functional, as expected, but unlike the coexpression of split constructs of a number of other membrane proteins, coexpression of the N- and C-terminal halves of SERT does not restore functionality. Nonetheless, whether expressed individually or together, these fragments produce a FRET signal. In addition, the expression of either fragment leads to the intracellular retention of full-length hSERT, similar to the findings described for DAT by Torres et al. (21). We thus infer that both the C- and N-terminal halves of SERT contain elements that support oligomerization. Using FRET donor photobleaching and a ß-lactamase protein fragment complementation assay, we have shown that constructs consisting either of the TM1 and TM2 domains or of the TM11 and TM12 domains, in each instance connected by the corresponding intervening extracellular loop, represent symmetrical intermolecular interaction sites for SERT (22). Whereas the interaction afforded by the TM1/TM2 domains may rely on the leucine heptad repeat (discussed above), the basis for the symmetrical interaction afforded by the TM11/TM12 sequence is unknown; a GxxxG motif in TM12 of SERT, not conserved in other NSS-family members, may, however, be critical for the latter. Given the evidence for two distinct symmetrical interaction sites in SERT, we propose, by analogy to the argument presented above for DAT (Figure 3⇑), that SERT is also arranged as a dimer of dimers (22).
Further Considerations of Quarternary Structure
Our ignorance regarding the tertiary structure of the NSS-family members makes it difficult to build a model that can explain all the data discussed above. In particular, the relationship between a TM2–TM2 interface, as predicted by mutagenesis studies in GAT (41), DAT (21), and SERT (22), and the symmetric TM4–TM4 and TM6–TM6 interfaces demonstrated by cysteine crosslinking, as well as with the symmetrical interaction involving the TM11/TM12 sequence in SERT, remains to be clarified. It is possible, for example, that the TM2 interface is shared with either the TM4 or the TM6 interface in a tetrameric complex, and ongoing studies attempting to crosslink cysteines of the TM2 of DAT may help to resolve this question.
It is important, however, to recognize that these results have been obtained in distinct transporters: DAT, SERT and GAT-1. Despite their high sequence similarity, these transporters differ substantially in their substrate and inhibitor specificity and targeting mechanisms. Thus, without further experimental evidence, it may be dangerous to generalize the implications of potential symmetrical or asymmetrical interaction domains. It is conceivable, for example, that TM4 and TM6 form the basis of symmetrical oligomerization interfaces in DAT but not in SERT, in which the symmetrical contact sites are instead in TM2 and the TM11/TM12 domains. Nonetheless, both of these arrangements would result in a dimer of dimers, which creates the possibility of an array-like quaternary structure, in contrast to the idea of the self-sustained oligomeric complex that was proposed by Klingenberg (9). Other potential arrangements may include asymmetrical contacts; this inference is based on our finding of interactions between different TM pairs of SERT using the ß-lactamase protein complementation assay: in these experiments, the TM5/6 pair was shown to interact with the TM11/12 pair (22).
The Biological Significance of Oligomer Formation
Although most (but not all) NSS-family members studied have been inferred to exist as oligomers, the role of oligomerization in transporter functionality remains enigmatic. As discussed above in the case of GAT-1, the replacement of leucine residues in TM2 with alanine abolishes FRET and leads to intracellular retention of the mutant proteins (41). Nevertheless, in vesicles prepared from human embryonic kidney cells expressing the mutant transporters, the leucine-to-alanine mutations affect neither affinity for GABA nor rate of transport (41). Given the recent evidence that DAT and SERT exist in the membrane as a dimer of dimers, it is tempting to conclude that the TM2 mutations in GAT-1 obliterate multimerization while preserving transporter activity and that the failure of surface expression reflects the loss of crucial intermolecular (GAT-1–GAT-1) interactions. We must, however, be cautious in interpreting the loss of FRET in our TM2 GAT-1 mutant proteins. The loss of FRET might indicate the presence of functional GAT-1 monomers, which would argue that oligomer formation is critical for surface targeting (see below) but not for transport activity. However, it is also possible that one oligomeric interface has been disturbed in the GAT-1 TM2 mutant, leading to loss of FRET but persistence of dimeric structure through a second interface, and in such a scenario one cannot conclude anything about the function of a monomer. Only by combining crosslinking and FRET studies can the proper interpretation be made, and these studies are ongoing in our laboratories.
Coexpression of different ratios of two differently epitope-tagged SERT variants, one sensitive and one insensitive to uptake inhibition by methanethiosulfonate compounds, has been used to argue for a functional interaction between the subunits of SERT oligomers (15). Furthermore, evidence for the functional oligomerization of human NET and DAT has been implicated by dominant negative mutations (20, 21). A precautionary note for the interpretation of dominant negative results, however, comes from the recent realization that nonfunctional DAT mutants [e.g., the human DAT Y335A mutant protein (50) ] can have unusual channel-like properties, and these properties may influence electrochemical gradients and thereby indirectly impair the function of the coexpressed wild-type transporter (A.-K. Meinild, H.H. Sitte, and U. Gether, unpublished results).
Evidence for and against the multimeric basis for transport functionality has been reported for other transport proteins. Quaternary structure appears to play only a modest role in controlling the transport cycle of bacterial transporters (10), and lactose permease, for example, has been shown to operate as a monomer (51). Band 3, the erythrocyte anion exchanger 1 (AE1), is a dimeric protein at the cell surface (52). However, inhibition of one subunit in the dimer does not affect the transport properties of the other subunit; thus, transport functionality resides completely within the protomeric subunit of the dimeric quaternary organization (53). The lack of dominant negative effects reveals no significant quaternary interactions of the wild-type glucose transporter 3 (GLUT-3), even when it is co-expressed in Xenopus oocytes with a threefold greater amount of a functionally inactive GLUT-3 mutant protein (54). Furthermore, biochemical studies reveal that although the members of the Na+/H+ -exchanger family are organized as oligomers, the minimal functional unit is the monomer (55). For the Na+/Pi -cotransporter, a similar conclusion was reached in an electrophysiological approach by coexpression of the wild-type transporter with a functionally distinguishable mutant variant (56).
Oligomerization in Sorting and Targeting
The localization of neurotransmitter transporters within the proper cellular compartment is critical for transporter function. The fact that mutation of the dimerization motif in TM6 of DAT, and that mutation of the leucine heptad repeat in TM2 of DAT and GAT, lead to intracellular retention suggests a role for oligomerization in the regulation of protein trafficking from the endoplasmic reticulum (ER) to the cell surface. This hypothesis is consistent with the observation of FRET early in the biosynthesis of neurotransmitter transporters.
In recent years, it has become apparent that NSS-family members are directed for transport through the cell by specific amino acid sequences. In the case of NET, basolateral localization in MDCK cells depends on a dileucine motif in the N terminus, although additional signals that specify targeting have also been proposed (57). Poyatos et al. (58) report that a similar dileucine signal is also implicated in the targeting of Glyt-1 to the somatodendritic compartment in neurons. The apical localization of GAT-3 in epithelial cells, on the other hand, is regulated by a binding motif for type-I PDZ domains, and fusion of the C-terminus of GAT-3 to GAT-2 redirects localization from the basolateral to the apical surface (59).
Whereas the studies discussed above focus on the polarized distribution of NSS proteins, very little is known about trafficking of these transporters early in the secretory pathway. Therefore, we will give a brief introduction to the topic of protein trafficking in the early secretory pathway as exemplified by other proteins, and we will attempt to relate these observations to the NSS-family members. In addition, we will present preliminary data that point to a possible role for oligomerization in the process of NSS trafficking.
After synthesis in the ER, proteins undergo a strict process of quality control before reaching their final destination (60). In general, proteins can emerge from the ER by one of three types of export: bulk flow, selective retention, or selective export. Bulk flow is observed for soluble proteins and is not relevant to transmembrane proteins. Selective retention indicates that the newly synthesized protein contains a retention signal and is thereby retained in the ER. Typical retention signals include the RXR motif (X indicates any amino acid), which occurs, for example, in the NR-1 subunit of the NMDA receptor. When the RXR motif of NR-1 is exposed at the protein surface, NR-1 is selectively retained within the ER; heterodimerization of NR-1 with NR-2 shields the retention signal, and functional heteromeric NMDA receptors are consequently trafficked to the cell surface (61).
The third mechanism, selective export, results from the presence of an ER export signal within the given protein’s primary structure. Di-acidic and dihydrophobic signals constitute the two major classes of ER export signals that have been identified. Di-acidic signals are important for the targeting of potassium channels (62) and the vesicular stomatitis virus glycoprotein (63). Examples of proteins that use dihydrophobic signals to escape the ER are ERGIC-53 (64) and some members of the p24 family (65). Unlike di-acidic signals, dihydrophobic signals are generally found at the extreme C terminus. ERGIC-53 and the p24 proteins that use dihydrophobic export motifs constitutively cycle between the ER and the Golgi apparatus. Recently, however, a divaline motif at the extreme C terminus was found to mediate ER export of the NR-1 subunit of the NMDA receptor (66) as well as of MHC-I.
The intracellular trafficking of proteins in eukaryotic cells is mediated by vesicular organelles. ER-derived vesicles are formed by a set of cytoplasmic proteins termed the coat protein complex II (COPII) (67) to capture cargo molecules for transport from the ER to the Golgi apparatus. The initial step in the formation of COPII vesicles is the activation of the small GTPase Sar1. Transfection of a dominant negative Sar1 variant into mammalian cells disrupts the export of all proteins tested and is thus deleterious (68). Active Sar1 associates with the Sec23/24 complex, and this tripartite complex forms the pre-budding complex. Sec23/24 in turn attracts the Sec13/31 complex to result in fully assembled COPII vesicles (see Figure 4⇓). In principle, a COPII vesicle composed of Sar1 and Sec23/24/13/31 is capable of budding from the ER. However, many additional proteins are recruited to the COPII vesicle, including several SNARE (for soluble NSF attachment protein receptor) proteins, which mediate fusion of the COPII vesicle with the target Golgi membrane (69). Moreover, distinct COPII vesicles can be specific for distinct classes of cargo proteins, and this diversity is achieved by a variety of cargo receptors. Recent work has identified two distinct cargo binding sites on Sec24, which might differentiate between dihydrophobic and di-acidic signals on cargo proteins (70). In addition to Sec23/24, many other cargo receptors have been reported to sort cargo molecules into COPII vesicles. Like Sec23/24, these cargo receptors also bind to dihydrophobic and di-acidic export signals. However, the cargo motifs that fully determine whether a cargo molecule will bind to a given cargo receptor remain undefined.
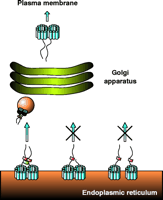
Trafficking of NSS proteins along the secretory pathway. This scheme shows the proposed model of NSS trafficking based on our results obtained with GAT-1. GAT-1 is synthesized in the ER and forms homo-oligomers. For simplicity, GAT-1 is shown as a dimer, although our recent data on DAT and SERT suggest NSS tetramers. The organization of the twelve membrane-spanning segments (blue) is unknown, and therefore the arrangement shown is merely for symbolic purposes. The ER export signal within the C terminus (shown as linear tail) of the transporter recruits the soluble Sec23/24 complex (red). Formation of a stable oligomer is proposed to bring the export signals with their Sec23/24 complexes into close proximity. This proximity in turn allows for the Sec13/31 complex (green) to bridge the Sec23/24 complexes, leading to the formation of a stable COPII coat and subsequent budding and transport of a COPII vesicle (orange sphere) to the Golgi apparatus. After modification in the Golgi apparatus, the NSS oligomers are targeted to the plasma membrane. The middle part of the figure illustrates the case in which a protomer in the oligomer lacks the export signal, so that no Sec23/24 complex is recruited and the assembly of a stable COPII coat is blocked; transporters are thus retained in the ER. The right part of the scheme illustrates the case in which either oligomer formation is inhibited or the interactions among protomers are weakened. In this case, each NSS protomer can still recruit Sec23/24 complexes, but subunits are too far away from each other for the Sec13/31 complex to bridge them efficiently. Under these conditions, formation of a stable COPII coat is not efficient and transporters are retained in the ER.
As discussed above, Scholze et al. (41) have shown that mutation of the GAT-1 TM2 domain results in proteins, defective in FRET and retained in the ER, that are nevertheless able to transport GABA. The functional transporter activity implies that the mutant proteins are correctly folded. Thus, oligomerization of GAT-1 per se appears to be essential for egress from the ER, and the processes of protein synthesis and folding are at least in part distinct from the process of ER export. From these data it is tempting to hypothesize that improperly oligomerized NSS proteins are selectively retained in the ER, even if otherwise properly folded. In such a scenario, a critical role of oligomerization might be to hide retention signals, in analogy to the findings obtained with the NR-1 subunit of the NMDA receptor (discussed above).
Recent preliminary findings indicate that selective export may be an important mechanism underlying the egress of GAT-1 from the ER (71 ; H. Farhan, O. Kudlacek, P. Scholze, V. Korkhov, M. Freissmuth, H.H. Sitte, unpublished results). Specifically, the C-terminal half of GAT-1 contains a putative ER export motif composed of three hydrophobic amino acids; deletions proximal to this export motif, as well as single amino acid substitutions within the export signal itself, lead to retention of the transporter in the ER. In addition, a GAT-1 mutant protein, disrupted within the leucine heptad repeat motif in TM2 and concomitantly deficient in FRET (41), is nevertheless able to interact with the COPII component Sec24 (H. Farhan, O. Kudlacek, P. Scholze, V. Korkhov, M. Freissmuth, H.H. Sitte, unpublished results). Although deficiency in FRET is not an absolute indication that this protein is deficient in oligomer formation, this mutant protein may represent a so-called “transition state,” in which newly synthesized transporters are capable of binding to COPII but not of promoting vesicle budding. These observations suggest the intriguing possibility that stabilization of the coat complex, which is important for COPII vesicle budding, may require an oligomeric assembly of the cargo transporters.
A similar scenario was recently demonstrated for the integral membrane protein Emp46p, which through hetero-oligomerization uses Emp47p as a cargo receptor (72). Emp47p uses a dihydrophobic export motif, which mediates binding to the Sec23/24 complex. An Emp47p mutant that is monomeric is still able to bind the Sec23/24 complex, but this binding does not suffice to promote efficient stabilization of the coat complex to initiate vesicle budding. Apparently, Emp47p must assemble into a homo-oligomer to support COPII vesicles competent of budding from the ER. Thus, although the monomeric variant of Emp47p is capable of binding to COPII, it is nevertheless retained in the ER. By analogy, we propose that oligomer formation of NSS proteins, apart from other potential functions, may serve to bring multiple export signals into close proximity in order to support assembly of the COPII coat (Figure 4⇑).
Conclusions
Although a large number of membrane proteins undergo oligomerization, the importance of this process is just beginning to be appreciated. One documented role for oligomer formation is to facilitate passage through the tightly controlled quality control system of the ER (60). Thus, as discussed above, oligomerization of membrane proteins appears to be essential for transport from the ER to the plasma membrane. Whether oligomerization of NSS proteins also plays a functional role in substrate transport is not yet clear; many other membrane proteins seem to be functionally competent as monomers, despite their participation in oligomer assembly.
As a determinant of the ultimate localization of neurotransmitter transporters at the cell membrane, and as a possible component of their transport function per se, the process of multimerization may in future be considered as a possible target of therapeutic intervention. As discussed above, the intermolecular crosslinking of DAT at Cys243, which is indicative of a TM4–TM4 dimeric interface, is dramatically inhibited by a high-affinity cocaine analog as well as by other uptake inhibitors (36). Conceivably, these inhibitors may thus function to prevent or to alter the conformation of intermolecular associations between DAT molecules; accordingly, disruption or modulation of the interaction of multimeric species may be a critical mechanism in the cocaine-induced inhibition of neurotransmitter uptake as well as of ion currents (73, 74). It is interesting to speculate whether the oligomeric interfaces might even contribute to the transport pathway or ion conduction pathways in DAT and related transporters, and this will merit further investigation.
If the mechanisms underlying oligomer formation in membrane proteins become understood in greater detail, our expanded knowledge might allow us to understand the potential role that protein–protein interactions might play in specific diseases. Since pathological states might arise from altered oligomerization of membrane proteins, it may be possible ultimately, through the design of small molecules that directly or indirectly alter protein–protein interactions, to counteract these detrimental processes and restore normal function to membrane proteins (75).
Acknowledgments
The authors wish to acknowledge support by the following granting institutions: Hygiene Fonds of the Medical University of Vienna, grant P14509 from the Austrian Science Foundation/FWF (to HHS); Grant 10507 from the Austrian National Bank (to HF) and NIH grants DA12408, DA11495, and MH57324 (to JAJ). We are grateful to Dr. Ulrik Gether for helpful comments on this manuscript.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Jonathan A. Javitch, MD, PhD, is Associate Professor of Psychiatry and Pharmacology in the Center for Molecular Recognition and in Physiology & Cellular Biophysics at the College of Physicians & Surgeons of Columbia University in New York. His laboratory focuses on understanding the structure and function of G protein–coupled receptors and neurotransmitter transporters.
Send correspondence to JAJ. E-mail jaj2{at}columbia.edu; fax 212-305-5594.

Harald H. Sitte, MD (left), Associate Professor of Pharmacology, and Hesso Farhan, MD (right), Research Fellow, work at the Institute of Pharmacology of the Medical University Vienna, Austria. Their research interests focus on the mechanisms that underlie oligomer formation and trafficking of neurotransmitter transporters and the repercussions of oligomerization in physiological and pathophysiological states.

