Histamine H3 Receptor Antagonists: Preclinical Promise for Treating Obesity and Cognitive Disorders
Abstract
The histamine H3 receptor is an attractive G protein–coupled receptor drug target that regulates neurotransmission in the central nervous system and plays a role in cognitive and homeostatic functions. Drug discovery efforts by numerous pharmaceutical companies have focused on the preclinical development of H3 receptor antagonists for the potential treatment of attention-deficit hyperactivity disorder, dementias, schizophrenia, as well as obesity and sleep disorders. This receptor exhibits molecular, pharmacological, and functional heterogeneity that informs the preclinical development of effective antagonists. Herein, we describe the biological and chemical implications for developing H3 receptor antagonists and their therapeutic potential as disclosed through animal models of cognition, sleep, and obesity.

Introduction
Histamine receptors have long been attractive drug targets, beginning with the development of classical antihistamines, such as diphenhydramine, desloratidine, and fexofenadine, which were directed at the histamine H1 receptor for the treatment of allergic reactions. The next drug class that was developed to block a histamine-mediated response comprises histamine H2 receptor antagonists such as cimetidine, ranitidine, and famotidine; blockade of histamine H2 receptors ameliorates gastric ulcers by inhibiting gastric acid secretion. Both histamine H1 and H2 receptor antagonists have been immensely successful therapeutic agents, benefiting millions of patients.
Since the original description of the histamine H3 receptor as a presynaptic autoreceptor that inhibits histamine release (1), along with its subsequent recognition as a heteroreceptor that regulates the release of other important neurotransmitters, there has been considerable effort by both academic and industrial laboratories to develop potent and selective H3 receptor antagonists (Figure 1⇓). Such antagonists are sought for the potential treatment of a variety of disorders affecting cognition (e.g., attention deficit and hyperactivity disorder [ADHD], Alzheimer’s disease, and schizophrenia), sleep (e.g., hypersomnia and narcolepsy), and energy homeostasis (e.g., obesity). The purpose of this paper is to review some of the preclinical considerations, including basic H3 receptor biology, as well as aspects of general drug discovery, that are relevant to the development of effective H3 receptor antagonists for the treatment of these diseases. In addition to the specific advances in H3 receptor antagonist research that we illustrate here, recent comprehensive reviews are highly recommended (2–5).
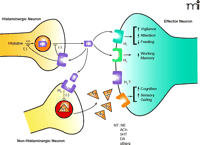
Schematic representation of H3receptor function. Stimulation of presynaptic H3 autoreceptors on histaminergic neurons by histamine (HA) inhibits the synthesis of histamine [through histidine decarboxylase (HDC)] and also inhibits the release of HA from the neuron. Similarly, stimulation of presynaptic H3 heteroreceptors on non-histaminergic neurons inhibits the release of a number of neurotransmitters (NT), including norepinephrine (NE), acetylcholine (ACh), serotonin (5HT), dopamine (DA), and others. These neurotransmitters can then activate their respective target receptors postsynaptically to evoke a variety of physiological responses as indicated in the figure.
The Histamine H3 Receptor: An Attractive CNS Drug Target
The histamine H3 receptor is a G protein–coupled receptor (GPCR) and one of four subtypes (H1, H2, H3, and H4) of the histamine receptor family. Since its cloning and characterization in 1999 (6), considerable progress has been made in the understanding of its structure, function, localization, and pharmacology. The H3 receptor consists of 445 amino acids and exhibits low similarity to other GPCRs, including the H1 and H2 receptors (~20% homology); it is most similar (~60% in the transmembrane domains) to the recently cloned histamine H4 receptor (2, 7). Additionally, alternative gene splicing can give rise to distinct H3 receptor isoforms, varying in length, N and C termini, sequence of the third intracellular loop, or reflecting deletions of transmembrane domains. To date, over twenty isoforms of the human histamine H3 receptor have been suggested by reverse transcription polymerase chain reaction (2, 7). The H3 receptor is localized primarily to the central nervous system (CNS), with highest expression, in rodents, in the cerebral cortex, hippocampal formations, striatum, and hypothalamus (8). Similarly in human, H3 receptor expression is prominent in the basal ganglia, globus pallidus, hippocampus, and cortex (9). Notably, many of these brain regions are critical for cognition (cortex and hippocampus) and sleep and homeostatic regulation (hypothalamus).
The limited expression of the H3 receptor in the periphery may prove fortuitous in terms of developing drugs that target areas of the CNS. Functionally, the H3 receptor also exhibits several properties that contribute to its attractive profile as a drug target. These include the ability to act as a presynaptic autoreceptor to modulate the release of histamine (10, 11), a neurotransmitter that plays a role in vigilance, attention, impulsivity, and feeding/weight regulation. Additionally, presynaptic H3 heteroreceptors modulate the release of other important neurotransmitters, such as acetylcholine (12), norepinephrine (13), dopamine (12), and serotonin (14), which are involved in cognition, mood, and sensory gating (Figure 1⇑). Thus, H3 antagonists may represent unique therapeutic modalities for the treatment of disorders of cognition, sleep, and energy homeostasis.
H3 receptors display a number of interesting pharmacological properties that differ among species, isoforms, and coupled signaling pathways, and these variations must be considered when designing H3 receptor antagonists. Prototypical H3 antagonists, such as thioperamide and ciproxifan, possess high affinity for the rat H3 receptor but have reduced (up to 100-fold lower) affinity for the human H3 receptor, so that their relative affinities for α2-adrenergic receptors and histamine H4 receptors can be problematic (15). Indeed, the first highly profiled non-imidazole H3 antagonist (A-304121), by Abbott, encountered this selectivity issue when it was found to be over 1,000-fold selective for the rat as opposed to the human H3 receptor (15). Subsequent work revealed the critical contribution of two amino acids in the third transmembrane domain to this species-dependent pharmacology (16, 17). Interestingly, the pharmacological profiles of H3 receptor ligands can also be dependent upon the G protein conformation and signaling pathway to which the H3 receptor is coupled; accordingly, functionally selective imidazole-based compounds such as proxyfan and GT-2331 can exhibit full agonism, partial agonism, or antagonism (18). [Chemical structures and ligand characterization are discussed below; consult Figure 3⇓ for chemical representations.]
Activation of the H3 receptor, coupled to Gαi/o proteins, engages a number of intracellular signaling mechanisms, including the inhibition of adenylate cyclase (6), activation of mitogen-activated protein kinase (MAPK) (8), and inhibition of the Na+/H+ exchanger (19). In addition to agonist-induced signaling, the H3 receptor is constitutively active and capable of signaling independently of agonist both in vitro and in vivo (20, 21). Most H3 antagonists synthesized to date possess inverse agonist activity, the magnitude of which may vary with the species, isoform, and signaling pathway being investigated (15, 22). Thus, most current therapeutic targeting of the H3 receptor is predicated on the design of a drug that not only blocks histamine agonism, but also functions as an inverse agonist and undermines tonic H3 receptor activity. However, no clinical data as yet have actually demonstrated the superiority of H3 receptor inverse agonists versus antagonists in blocking an agonist response.
Given the complexities and variabilities discussed above, careful consideration must be made with respect to characterizing and optimizing H3 receptor antagonists during preclinical development. Pharmacological heterogeneity that arises from species and isoform differences in H3 antagonist binding potencies, agonist functional potencies, potential receptor dimerization, constitutive activity, and differential coupling mandates a research strategy that allows for meaningful interpretation across species and for translation into the clinic. Only the thorough interrogation of antagonists against multiple H3 receptor isoforms and in different functional signaling assays can assure compound efficacy and support drug discovery strategies.
The H3 Receptor in Sleep Disorders
Sleep disorders such as narcolepsy, hypersomnia, and daytime somnolence due to jet lag or other sleep disruptions can have adverse consequences on normal daily functioning but may be amenable to pharmacological intervention. Current therapy includes drugs such as modafinil (Provigil®), which promotes vigilance and increases histamine release in the hypothalamus (23). In fact, a considerable body of evidence supports the role of histaminergic neurons in maintaining wakefulness (5, 24). Histaminergic neurons arising from the hypothalamic tuberomammillary nucleus project to a variety of brain regions, including the cortex, preoptic and anterior hypothalamus, thalamus, and brainstem, that regulate the sleep–wake cycle (5, 24). Additionally, extracellular histamine levels in the frontal cortex are positively correlated with the amount of wakefulness in rats (25), and stimulation of postsynaptic H1 and H2 receptors facilitates cortical activation (26). Indeed, histaminergic involvement in the sleep–wake cycle has been exploited clinically by the use of H1 antagonists such as diphenhydramine and hydroxyzine to facilitate sleep. Thus, preclinical studies as well as clinical results strongly support the role of histamine in the regulation of sleep.
H3 receptors have received considerable attention as potential targets for the control of arousal and vigilance as well as for the treatment of sleep disorders, because they colocalize with histaminergic neurons in brain regions that regulate the sleep-wake cycle and they modulate histamine release and levels in the CNS (5). Researchers have successfully used a variety of selective H3 agonists and antagonists as pharmacological tools to study sleep in a number of animal models. The administration of selective H3 receptor agonists, such as R-α-methylhistamine (27, 28), BP-294 (29), and SCH50971 (30), increases sleep time and slow wave sleep in cats and rodents (27–29) and produces sedation in the guinea pig (30). Work with selective H3 antagonists provides an even stronger case for the role of H3 receptors in arousal; H3 antagonists such as thioperamide increase wakefulness in cats and rats (27, 28) and decrease slow wave sleep and REM sleep in rats (28). Similarly, ciproxifan depresses cortical slow wave activity and enhances fast rhythms, eliciting wakefulness in cats (10). Interestingly, H3 receptor antagonists such as GT-2331 (31) and JNJ-10181457 (32) decrease the number and duration of narcoleptic episodes in a canine model (i.e., Dobermans bearing a genetic defect in the orexin pathway). Additionally, mice in which either the H3 receptor itself (33) or histidine decarboxylase (11) has been knocked out are insensitive to the wake-promoting effects of H3 receptor antagonists. Although no clinical efficacy of H3 antagonists has yet been presented, the future appears promising for the utility of these agents in the treatment of sleep disorders.
The H3 Receptor in Obesity
Obesity is a worldwide health crisis, attributable to poor diet and physical inactivity, that contributes to a number of pathophysiologic conditions including hypertension, coronary heart disease, diabetes, and stroke (4, 34). Despite concerted efforts within the pharmaceutical industry, there are only two drugs approved for long-term treatment of obesity, with a third awaiting FDA approval. Each of the three drugs works by a different mechanism, reflecting the complex etiology of the disease. Sibutramine (Meridia®) is a non-selective norepinephrine/serotonin/dopamine reuptake inhibitor that manifests appetite suppressant and satiety enhancer activity; orlistat (Xenical®) is a lipase inhibitor that inhibits intestinal fat absorption; and rimonobant (Acomplia®) is a CB1 cannabinoid receptor antagonist that shows promising weight loss effects in clinical trials.
Histamine has long been known to play a critical role in homeostatic regulatory functions such as control of food intake and maintenance of body weight in animals (4). Histamine-synthesizing neurons in the tuberomammillary nucleus (see above) project to the paraventricular nucleus and ventromedial hypothalamus, two hypothalamic regions known to regulate food intake (35, 36). Masaki and co-workers have demonstrated that central infusion of histamine reduces fat accumulation, upregulates uncoupling protein, decreases leptin, and improves insulin sensitivity in a leptin-resistant mouse model of obesity (37). Additionally, postsynaptic H1 receptor activation in the ventral/medial hypothalamus has been shown to suppress food intake in rats (38, 39). Interestingly, weight gain associated with the use of typical and atypical antipsychotics has been suggested to be the result of H1 receptor blockade (40).
A number of investigational H3 antagonists have been evaluated for anti-obesity effects in a variety of animal models (4). Early work with the prototypical imidazole-based H3 receptor antagonist thioperamide showed that indeed appetite and/or the amount of food consumption was decreased (41, 42) although a later study (43) indicated that some of the effects seen with the peripheral, but not central, administration of thioperamide may have resulted from taste aversion. Other imidazole-based H3 antagonists, including GT-2394, reduce cumulative food consumption and weight gain in Sprague Dawley and obese Zucker rats (44). Clear insights from the creation of H3 receptor knockout mice are still forthcoming, with reported phenotypes ranging from slightly decreased body weight (33) to increased weight-gain rates (45). The authors of the latter study suggested that the slightly obese phenotype might result from the desensitization of postsynaptic H1 receptors (45). Interestingly, thioperamide did not decrease an acute feeding response in H3 receptor knockout mice (45).
More recently, efforts to develop an H3 receptor antagonist anti-obesity agent have focused on non-imidazole–based compounds in order to avoid potential drug–drug interactions. Novo Nordisk has extensively investigated the anti-obesity activity of two potent and selective non-imidazole H3 antagonists (NNC 38-1049 and NNC 38-1202). Upon acute administration, NNC 38-1049 significantly increases extracellular histamine concentrations in the hypothalamus and reduces food intake in a dose-dependent manner (46). Similarly, NNC 38-1202 elevated hypothalamic histamine levels acutely and induced weight loss upon daily administration to obese rats and primates (47). Abbott Laboratories has described the anti-obesity effects of several potent and selective non-imidazole H3 receptor antagonists. A-331440 causes dose-dependent weight loss in a diet-induced obese mice model and prevents weight gain in genetically obese (ob/ob) mice (48); weight loss in the diet-induced obese mice, which was largely specific for body fat, was associated with reductions in food consumption and plasma leptin levels (48). A closely related analog, A-423579, also effectively reduces weight gain in diet-induced obese mice and rats, with concomitant decreases in fat content, plasma leptin levels, and triglycerides (49). Interestingly, not all H3 receptor antagonists with potent in vitro activity also display potent anti-obesity activity, including JNJ-5207852 (50) and A-631972 (51) although the reasons for this have not been determined.
The H3 Receptor in Cognitive Disorders
Cognitive Domains
Cognitive function is disturbed across a broad range of neuropsychiatric diseases encompassing patients of all ages. ADHD, which can affect up to 7 % of the pediatric population, can also continue into adulthood. Alzheimer’s disease (AD) and its precursor, mild cognitive impairment, are associated with cognitive decline during mid to late life, which is of increasing public health concern as populations age. Schizophrenia is a neurocognitive disorder and was originally characterized as such, despite a focus on treating positive symptoms in recent years. This disorder is characterized by severe cognitive dysfunction that becomes apparent during adolescence and persists throughout life. (Down syndrome, Parkinson’s disease, and fetal alcohol syndrome are also characterized by cognitive dysfunction; however, these will not be reviewed here.) Although various drugs have been employed to treat cognitive impairment (e.g., stimulants for ADHD, cholinesterase inhibitors for AD, and antipsychotics for schizophrenia), efficacy is generally incomplete, and side effects are common.
Most treatment strategies for ADHD, AD, and schizophrenia have focused on a single neurotransmitter system. For example, some symptoms of ADHD are amenable to stimulants such as methylphenidate, which increase dopamine levels. In AD, acetyl-cholinesterase inhibitors such as donepezil show transient and modest efficacy. D2-dopaminergic and 5-HT2-serotoninergic receptor antagonists are effective at treating positive symptoms of schizophrenia but do not robustly improve cognitive impairments. In each of these disorders, however, it is likely that disruption of multiple neurotransmitter systems is involved. H3 receptor blockade, able to elevate concentrations of histamine, acetylcholine, dopamine, serotonin, noradrenaline, and glutamate, may thus offer a means for targeting cognitive processes, which often rely on the integration of multiple neurotransmitter systems. Early reports of improved cognitive function with H3 receptor ligands were unfortunately not uniformly consistent, varying with behavioral tests and ligands used, and likely reflecting the interspecies and molecular heterogeneity of H3 receptors or the specificity of the ligands used. More recent work has focused on the use of highly selective antagonists in selected animal models that involve particular cognitive domains. Below, we have attempted to organize the various cognitive domains that are readily modeled in animal experiments and that are relevant to the treatment of ADHD, AD, and schizophrenia. In particular, we summarize a system that conveys the reported efficacy of H3 receptor antagonists and reflects various tests that are used in our laboratories (Figure 2⇓).
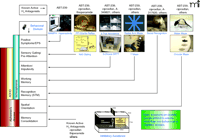
H3receptor antagonists exhibit broad efficacy in preclinical models of cognition. Animal models depicted across the top are used to analyze the cognitive behavioral domains (indicated by connecting arrows to respective domains in left column) thought to be important for human diseases such as ADHD, Alzheimer’s Disease, or schizophrenia (53). The animal models where H3 antagonists have been shown to be efficacious include (from the left): 1) reduction of methamphetamine (MAMPH) hyperactivity; 2) enhancement of prepulse (PPI) inhibition of startle and N40 gating responses; 3) 5-trial inhibitory avoidance and 5-choice serial reaction time test (SRTT); 4) radial arm maze and Y maze; 5) social recognition; 6) water maze and circular maze; and 7) inhibitory avoidance. The indicated efficacious H3 receptor antagonists, such as ciproxifan and ABT-239, that are shown above each animal behavioral model are not intended to constitute a comprehensive list of all H3 antagonists tested in these models but to illustrate the broad efficacy of H3 receptor antagonists across cognitive domains.
Memory Consolidation
Memory consolidation may be defined simply as the conversion of recent learning experiences into long-term memory, impairments of which are apparent particularly in AD. The inhibitory avoidance test (Figure 2⇑), a standard behavioral probe of learning, has revealed that thioperamide improves recall in a mouse model of premature senescence (52) as well as in spontaneously hypertensive rat pups (53). Thioperamide and clobenpropit can also prevent scopolamine-induced amnesia (54), whereas H3 agonists can reduce ACh levels and diminish cognitive performance (55). Further, H3 receptor knockout mice are insensitive to the effects of scopolamine in an inhibitory avoidance paradigm (33), supporting a role for H3 receptor modulation of cholinergic function in memory acquisition. On the other hand (56) another report failed to demonstrate cognitive enhancement with thioperamide.
Spatial Orientation
Spatial memory is the recording of local information for navigation of the environment. Impairments in spatial orientation are typical of AD. We have reported that ciproxifan and thioperamide can reduce scopolamine-induced errors in discriminating the location of a fixed, compared to a floating, escape platform using a two-choice discrimination version of the water maze (57, 58). Similar results are seen with ABT-239, which shows dose-related reversal of scopolamine-induced deficits in the two-platform water maze. In Barnes maze studies, mice lacking H3 receptors learn spatial cues more aptly than do wild-type mice (59), consistent with the concept that blockade of H3 receptors should enhance spatial reference memory. In contrast, the H3 receptor agonist R-α-methylhistamine also reduces scopolamine-induced deficits in the Morris water maze (60, 61).
Social Recognition (Short-Term) Memory
Impairments in social recognition memory are apparent in AD, but may also be relevant to social (transient) cognitive impairment in schizophrenia and ADHD. The social recognition test in rats relies on the retention of memory through which an adult animal uses olfactory cues to recall a social interaction with a conspecific juvenile. Significantly, the test has been used to show that the administration (intracerebroventricular; i.c.v.) of selective histaminergic agonists enhances social memory, whereas recall is disrupted by the inhibition of histamine synthesis (62). In particular, thioperamide (50) as well as several non-imidazole H3 receptor antagonists, such as A-304121, A-317920, and ABT-239 (12, 63) have been attributed with pro-cognitive effects. ABT-239 appears in fact to endow aged rats with levels of social recognition that are comparable to those of younger rats; the potential relevance of these results to AD is intriguing (12).
Working Memory
Working memory, which is impaired in AD, ADHD, and schizophrenia, may be defined simply as memory for and attention to items currently being processed. Examples of relevant tests include variations of the water maze, Y maze, radial arm maze, and tests of delayed match-to-sample memory. Increased error rates in the radial maze following disruption of histamine synthesis have been correlated with declining histamine content in both cortex and hippocampus, and these deficits can be reversed by either i.c.v. histamine or thioperamide (64); scopolamine-induced deficits in maze performance can be similarly reversed (65, 66). Interestingly, clobenpropit also reverses radial arm maze deficits induced by MK-801 (dizocilpine), a non-competitive glutamatergic antagonist, suggesting the potential utility of H3 antagonists in psychotic disorders (67). In Y-maze tests, thioperamide reverses scopolamine-induced deficits in a mechanism that may involve central histamine H2 receptors (68), in contrast to numerous other studies indicating H1 receptor involvement in cognitive aspects of H3 receptor blockade.
Attention and Impulsivity
Whereas attention is impaired in AD, ADHD and schizophrenia, impulsivity is a particular problem in ADHD. Examples of tests effective at assessing both attention and impulsivity include the repeated acquisition passive (inhibitory) avoidance model and the five-choice serial reaction time test. In the standard passive avoidance test, trials are separated generally by twenty-four hours, so that long-term memory can be evaluated. In the repeated acquisition version, several trials, typically separated by minutes, allow evaluation of aspects of attention and learning. When this test is used with spontaneously hypertensive rat pups, impulsivity that may be relevant to ADHD can be assessed (69), and in fact both methylphenidate (used clinically to treat ADHD) and ABT-418 [a nicotinic agonist that showed positive effects in a small clinical trial for ADHD (70)] are efficacious at clinically relevant exposure levels (Hancock and Fox, unpublished observations). In naïve rats, several compounds from Gliatech appear to improve working memory (71), whereas thioperamide, ciproxifan, and GT-2331 are also efficacious in spontaneous hypertensive rat pups (69, 72). Non-imidazole H3 antagonists such as A-304121, A-317920, A-349821 (63, 73), as well as ABT-239 and several analogs, also work in this model of ADHD (74). Unfortunately, far fewer studies have examined the effects of H3 receptor antagonists in the five-choice serial reaction-time test. This test requires sustained attention over time, and is frequently used to evaluate compounds acting on nicotinic cholinergic receptors (75). Given the increased interest in ADHD and the potential for H3 receptor antagonists to improve attention and impulsivity, it is anticipated that new data with H3 receptor antagonists, especially generated using the five-choice serial reaction time test, will be forthcoming.
Schizophrenia: Sensory Gating and Psychosis
Sensory gating, defined simply as the filtering or gating (at a “reflex level) of extraneous stimuli, is perhaps best understood through the pre-pulse inhibition (PPI) test. In normal humans and mice, the startle response to a loud acoustic stimulus is attenuated if preceded by a “preemptory” acoustic stimulus of lower magnitude (i.e., the pre-pulse). Schizophrenia patients and model mouse strains, however, show no attenuation of the startle response after the pre-pulse. Indeed, antipsychotic drugs can enhance the ability of the pre-pulse to inhibit the startle response. Sensory gating may also be relevant to ADHD and AD. Intriguingly, thioperamide, ciproxifan, and ABT-239 can attenuate the deficits of pre-pulse inhibition in model mice to the same degree as can antipsychotic compounds (12 ,76). In addition, ABT-239 normalizes auditory responses to paired “clicks” or tone pulses in the N40 waveform model of sensory gating that relies on auditory evoked potentials recorded directly from the hippocampus in DBA/2 mice (12).
Given that a variety of dopaminergic agonists can induce psychotic symptoms (77), methamphetamine-induced hyperlocomotion is a behaviorally relevant model for psychosis. Methamphetamine-induced hyperlocomotor activity can be attenuated by ciproxifan in mice (78), as well as by the antipsychotic drug risperidone and the H3 receptor antagonist ABT-239 (12). Interestingly, risperidone elicits a notable sedative effect prior to the administration of methamphetamine, which is not observed with ABT-239 (12).
H3 Receptor Antagonists: Design Strategies
R-α-methyl histamine, (Figure 3⇓, compound 2; RAMH) has long been used in vitro and in vivo as a potent and highly H3 receptor–specific full agonist (79 ,80); NAMH (compound 3) similarly has found use in binding assays as a potent agonist radioligand. Other synthetic agonists include the more recently reported methimepip (compound 4) (81). The available histamine H3 receptor agonists contain an imidazole moiety, as do ciproxifan (compound 5) (10, 82) and thioperamide (compound 6) (20, 83). These compounds have been shown to block agonist dependent effects in vitro (e.g., by fluorescent imaging) and ex vivo, and have shown inverse agonism in a number of assays (e.g., GTPγS binding).
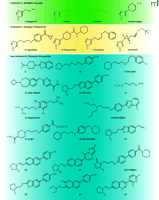
Chemical structures of H3receptor ligands. Agonists (green box), imidazole H3 receptor antagonists (yellow box), and non-imidazole H3 receptor antagonists (blue box) are indicated. Compounds 7 and 8 exhibit functional selectivity (agonism, partial agonism, and antagonism) dependent upon the assay system investigated. See text for details.
Standard H3 antagonists elicit a spectrum of in vivo effects, such as diminution of RAMH-induced dipsogenia, enhancement of social memory, and avoidance acquisition learning in spontaneous hypertensive rat pups thought to be a model of ADHD (63a, 63b, 84). In contrast, other imidazole-containing analogs show more complex, assay-dependent pharmacology. Proxyfan (compound 7) has been nominated as fulfilling the conditions of “protean agonism,” because it manifests a full range of receptor interactions (i.e., agonism, antagonism, and full inverse agonism), depending on assay conditions (85). Another example of the potential for complex pharmacology of the imidazole group is GT-2331 (compound 8), formerly a Phase I candidate compound (apparently not advancing further). This compound has been reported to show agonist-like effects in some systems and antagonist-like effects in others (86). In addition to the potential for complicated H3 receptor–mediated pharmacology, there are additional concerns when considering the suitability of lipophilic imidazole-containing compounds as potential drugs. Drugs that bear an imidazole group show high potential for cytochrome P450 (CYP) inhibition and may contribute to drug–drug interactions by slowing the metabolism of co-administered drugs (87). Imidazole-containing compounds have also been reported to have less efficient CNS penetration and may thus fail ultimately to reach their target. Because of such well recognized challenges, almost all recent research directed toward development of H3 antagonist ligands has been directed toward the so-called “non-imidazoles” (Figure 3⇑).
The field of non-imidazole H3 antagonist ligands is today characterized by an extremely broad diversity of structural classes, with structural diversity increasing over time, rather than converging toward any common group of pharmacophores. This trend reflects the ingenuity of investigators and the large number of teams working toward finding H3 antagonists, but may also indicate an inherent ability of the receptor to accommodate a wide variety of pharmacophores. The initial disclosures and explorations of non-imidazole H3 antagonists were from a variety of institutions. Isopropylpiperid inylbenzothiazoles (Figure 3⇑, compound 9) were reported as early non-imidazole H3 antagonists (88), admittedly of rather low potency (pA2 7.03) by today’s standards of nanomolar and subnanomolar potencies. The first reports of non-imidazole H3 ligands [compound 10; (89)] uncovered important SAR principles that were used in the design of later antagonists. It was found with these early molecules [e.g., compound 11 (90, 91)] that the imidazole group was not necessary for activity at H3 receptors, but that a basic amine (almost always a tertiary amine) was a requirement. Demonstrations of utility with model compounds in a number of animal models of human disease, including asthma and allergic rhinitis, dementia, cognitive and learning disorders, and narcolepsy, have motivated the organized search for highly drug-like H3 antagonists (3). Teams from JNJ, Schering, Abbott, GSK, Pfizer, UCB Pharma, Merck, Banyu, Lilly, Sanofi-Synthelabo, Roche, and other industrial and academic groups have contributed to the growing number of non-imidazole H3 ligands with nanomolar in vitro potency at human and animal H3 receptors. A number of high-profile H3 antagonists have been reported recently, with some nominated as potential clinical candidates. Of particular recent note is JNJ-17216498, a compound directed to treating narcolepsy, and reported to be “well tolerated and to possess good pharmacokinetic properties” in humans (92); the exact structure and properties are not reported. Another recently described compound is GSK-189254 (compound 12), targeted toward treatment of dementia (93). The compound is extremely selective and potent at H3 receptors, has excellent pharmacokinetic properties in rat and monkey, and is efficacious in an animal model of dementia. Additional highlighted histamine H3 antagonists include ABT-239 [compound 13; (51)] and ABT-834 (structure and properties not described), targeted toward cognitive disorders and ADHD. Rather than reviewing the large body of research, we touch here on highlights in the evolution of H3 antagonist design, focusing on research from our own laboratory.
In order to find a candidate suitable for Phase I, dozens of independent parameters and properties must be optimized and evaluated. If the process of drug discovery can be said to be a hurdler’s race, then it is also a race where the racer must return to the starting gate after booting even a single hurdle. Key hurdles to the development of an H3 antagonist include assessment of the antagonist/inverse agonist potency across multiple species (e.g., rat, mouse, and monkey), and the potential for off-target effects. Selectivity, pharmacokinetic profiles, and toxicology assessments must be made in multiple species, and the potential for drug–drug interactions in humans should be minimized, with the compound neither inhibiting human metabolic enzymes nor being exclusively dependent on any single enzyme for its own clearance. Candidates should have high projected therapeutic indices, as assessed in preclinical tests of acute toxicity, particularly cardiovascular (e.g. hemodynamics and QTc potential), respiratory, and gastrointestinal effects.
Within the diverse H3 antagonist literature, one pharmacophore class that appears is the amine-propyloxyphenyl function (compound 14). Descriptions of molecules bearing this pharmacophore have emerged from diverse sources: the early design optimization work of Ganellin (93); the Bioprojet group [compound 11 and related analogs (90, 91)]; from a marine natural product aplysamine (compound 15) (94); and from high-throughput screening efforts [e.g., RWJ-20085 (compound 16)] (95) and A-923 (compound 17) (96).
Whereas non-imidazole antagonists must have at least a single basic amine (thought to interact with Asp114 in the H3 receptor) (17), a second basic amine can often boost potency, as is exemplified by both JNJ-5207852 (compound 18), a wake-promoting H3 antagonist (50), and a dibasic analog of ABT-239 [compound 19); Ki = 100 pM for the human H3 receptor] (97). This potency-enhancing effect may arise from a beneficial interaction of the second basic amine with an acidic or hydrogen-bonding residue in the receptor. Caution must be exercised, however, and care taken to assess the dibasic molecules for their propensity to induce phospholipidosis, a type of toxicity that is proposed to occur when molecules partition into lipid bilayers and impair normal phosholipid turnover (98). Indeed, compound 19 proves to be a much more potent inducer of phospholipidosis relative to its monobasic congeners.
One design principle that has sometimes been used in the H3 antagonist field is the principle of enhancing the rigidification of the molecular skeleton (99). There are empirical reports that reduction in the number of rotatable bonds is associated with enhancement in overall drug-likeness. Another aspect is that conformationally constrained ligands of a target receptor should have a lower probability of interacting with off-target receptors. In practice, however, the majority of molecules bearing additional conformational constraints are more difficult to synthesize, and many of the new structural classes will be inactive, because there will also be fewer favorable conformations for binding to the target receptor. Nevertheless, the benzofuran ABT-239 (compound 13) was designed by rigidification of an amine-propyloxyphenyl pharmacophore (51, 99). The high oral bioavailability and good pharmacokinetics seen with the benzofuran-containing class of compounds (53% oral bioavailabilityand t1/2 = 5.3 hours) are also found in additional series of bicyclic heterocycles, such as in the naphthylene (compound 20; 55% oral bioavailabilityand t1/2 = 5.5 hours) and quinoline (compound 21; 90%oral bioavailability t1/2 = 5.3 hours) series of compounds (100, 101).
The overall lipophilicity of a molecule, expressed as the measured logD and ClogP, plays an especially important role in the suitability of a molecule as a CNS drug candidate. For example, although A-349821 (compound 22) and ABT-239 (compound 13) show almost identical rat H3 receptor potency, the latter, being the more lipophilic, distributes into the brain much more significantly than the former after equivalent dosing (51). Possibly in part due to its higher brain penetration, ABT-239 was found to be 10- to 100-fold more potent than A-349821 (compound 22) in a key in vivo efficacy model. Compounds that are very highly lipophilic, however, are also more likely to associate with plasma proteins and cell membranes, reducing the amount of “free drug” available for binding to the target receptor.
For other off-target properties (e.g., metabolic stability), there are not always clear-cut structure–activity relationships to guide compound design. For this reason, a library of compounds that represent structural diversity can be beneficial, and additional heterocyclic analogs of ABT-239 (102) and other molecules warrant investigation. Compounds 23, 24, and 25 illustrate opportunities for additional examples of novel H3 antagonist ligands.
Concluding Remarks
Since its cloning, considerable progress has been made in understanding the properties of the H3 receptor as well as in developing H3 antagonists. There is increased appreciation for the complex biology associated with this receptor, particularly for the heterogeneity of its molecular properties (i.e., isoforms, oligomerization, localization), pharmacology (i.e., species- and isoform-dependent potency and efficacy) and functional properties (i.e., constitutive activity, isoform-dependent signaling, and functional selectivity). Despite this complexity, both academic and pharmaceutical laboratories have synthesized a large number of highly potent and selective H3 receptor antagonists, with efficacy in a variety of preclinical animal models of cognition, sleep, and obesity. However, no clinical data for an H3 receptor antagonist are yet available, perhaps reflecting the multiple hurdles related to H3 receptor biology as well as meeting the challenges of producing drug-like molecules. Further understanding of the complex biology of this receptor is clearly mandated, however, given the potential for treating a number of important human diseases.
Acknowledgments
The authors want to thank the many Abbott members of the extended H3 Receptor Antagonist Project team for their considerable contributions to the work presented in this review that flowed from their collaborative effort. The authors also wish to thank the contributions of Dr Kaitlin Browman and Dr Isabel Lopez to the schematics.
Footnotes
-
The authors wish to dedicate this review to the memory of Arthur A. Hancock (1946–2005) who led the H3 Receptor Antagonist Project at Abbott Laboratories until his unfortunate passing last year. He is missed greatly.
- © American Society for Pharmacology and Experimental Theraputics 2006
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63a.↵
- 63b.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵

Marlon Cowart, PhD, author or inventor of more than 80 papers and patents, is a Volwiler Associate Research Fellow and senior group leader of the medicinal chemistry research team working on histamine H3 receptor antagonists at Abbott Laboratories, in the Department of Neuroscience Research.

Gerard B. Fox, PhD, is the Project Leader of Experimental Imaging in Global Pharmaceutical Research and Development at Abbott Laboratories in Advanced Technologies. He is the author of more than 55 papers and book chapters.

Timothy A. Esbenshade, PhD, is a Senior Group Leader in Global Pharmaceutical Research and Development at Abbott Laboratories in the Department of Neuroscience. Please address correspondence to TAE. E-mail: Tim.Esbenshade{at}Abbott.com; fax 847-937-9195.

