Immunodeficiency Is A Tough Nut to CRAC: The Importance of Calcium Flux in T Cell Activation
The immune system is composed of a complex armamentarium of diverse cell types that act in concert to protect the body from pathogenic organisms and foreign particles. The multi-component nature of the immune system enables exquisite control of highly regulated, fine tuned responses; however, this feature of the immune system also provides numerous opportunities for problems to occur. Although overresponsiveness by specific arms of the immune system can be life threatening—for example, T cell activation that arises from autoimmune reactions—frequently, overresponsiveness can be held in check through the administration of immunosuppressing therapies [for example, see (1)]. It is much more likely that the inability to raise a sufficient immune response to a pathogen will have life-threatening consequences. In particular, defects in the development of T lymphocytes not only impair normal immune function but can lead to primary immunodeficiency (PID).
PIDs are relatively rare and occur in approximately 1 in 2,000–10,000 live births (2). The consequences of PID depend on the immune system component involved and the number of immune components affected. Accurate data on the development of PID are difficult to obtain because infants and young children may die owing to infection without accurate diagnosis of their condition (3). This also holds true for severe combined immunodeficiency (SCID), a particular subset of PID that is estimated to occur in 1 in 50,000–100,000 live births (3).
SCID is a disease caused by a heterogeneous group of genetic disorders that are characterized by the inhibition of T cell differentiation and may also include the abnormal development of B cells and NK cells (4, 5). Thus, the primary classification of SCID can be made by grouping patients who have B cells and those who do not have B cells, followed by noting the presence of absence of NK cells (5). Further subset categorization, made on the basis of cellular and metabolic dysfunctions, has led to the classification of five distinct SCID patient groups: 1) defects in cytokine receptors and cytokine signaling (Table 1⇓); 2) defects in purine pathway enzymes leading to the loss of lymphocyte function; 3) defects in the recombination of antigen receptor genes of B cells and T cells; 4) defects in other lymphocyte specific genes (Table 2⇓); and 5) SCID phenotype arising as component of other syndromes.
The most frequently occurring form of SCID is X chromosome–linked SCID (X-SCID), which accounts for approximately 50–60% of all SCID cases (5), followed in prevalence by adenosine deaminase–deficiency SCID (ADA-SCID), which arises from a defect in purine metabolism and includes 15–20% of all cases (5, 6). Other common genetic defects that lead to SCID include mutations in RAG1, RAG2, or both (resulting in defective rearrangements of the T cell receptor and the B cell receptor), accounting for approximately 20% cases (7).
Rarer forms of SCID have been identified in patients (7). Among these forms are major histocompatibility complex (MHC) class I expression deficiency arising from mutations in TAP1 and TAP2 (genes whose products are important for transport of MHC class I molecules to the cell surface); MHC class II deficiency arising from mutations in genes encoding the transcription factors Class II Transactivator (CIITA), Regulatory Factor X 5 (RFX5), Regulatory Factor X Associated Protein (RFXAP), or Regulatory Factor X Ankyrin-containing protein (RFXANK); defective T cell receptor complexes arising from mutations in genes encoding various polypeptides that comprise the complex (i.e., ZAP70, CD3D, CD3E). It is currently understood that SCID can arise from mutations in at least ten different genes (8), and there are likely more genes involved in this disease which remain to be discovered.
A newly uncovered mutation has recently been identified in SCID patients that results in defective T cell signaling. This mutation is located within ORAI1, a gene encoding a component of the Ca2+ release–activated Ca2+ (CRAC) channel. To date only a few SCID patients have been described with a T cell–restricted Ca2+ flux defect, nonetheless, this new genetic defect must now be considered when a diagnosis of SCID is made (2, 9, 10).
Patients with ORAI1 mutations present with no T cell signaling capabilities; therefore, they do not mount a normal immune response. In healthy individuals, T cells are key to the adaptive immune response and under normal circumstances, after T cell receptor activation, a signaling cascade ensues that culminates in the phospholipase Cγ1-mediated hydrolysis of phosphatidylinositol-3, 4,5-bisphosphate into diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3) (Figure 1⇓). IP3 binds to the IP3 receptor in the membrane of the endoplasmic reticulum (ER) initiating the release of Ca2+ from stores within the lumen of the ER. The depletion of Ca2+ from the ER triggers the opening of the CRAC channels in the plasma membrane resulting in Ca2+ influx and elevated intracellular Ca2+ levels (11, 12). This process is essential for T cell activation and proliferation because it results in Ca2+-dependent calcineurin-mediated dephosphorylation of nuclear factor of activated T cells (NFAT), leading to NFAT translocation to the nucleus and transcription of various genes essential for T cell effector function. The activities of the transcription factors activator protein–1 (AP-1) and nuclear factor–κB (NF-κB) are also modulated by Ca2+ levels; indeed, 75% of all activation-regulated genes of T cells show dependence upon Ca2+ influx through the plasma membrane via CRAC channels (13). Thus, calcium transport is essential for the initiation of transcription of genes that drive T cell proliferation and effector function, and CRAC channels are the major pathway for Ca2+ influx into T cells following TCR triggering. Therefore, it is not surprising that a mutation in a gene that is proposed to be an essential component of the CRAC channel will result in a form of SCID. In T cells from patients with such a mutation, the release of Ca2+ stores from the ER following T cell activation fails to trigger Ca2+ store–operated Ca2+ entry into the cells through the CRAC channels in the plasma membrane. Therefore, despite normal Ca2+ release from the ER stores, T cell activation is impaired because of a complete failure of CRAC channel opening resulting in almost no Ca2+ influx into the cells and consequently impaired NFAT-dependent gene activation.
The molecular constituents of CRAC channels have yet to be fully elucidated and have been the subject of extensive research for many years (14). To date, little is known about the activation of the CRAC channel or the molecular basis of its activity. Numerous genes and mechanisms have been suggested to explain CRAC channel activity, for example the Transient Receptor Potential (TRP) family of ion channels, but evidence supporting their role has been inconsistent (15, 16). Recent research, however, has suggested that Orai1 may be a subunit or key component of the CRAC channel complex (2, 17, 18). Depletion of Orai1 by RNA interference greatly suppresses Ca2+ influx and CRAC channel activity (18). Orai1 is thought to work in concert with stromal interaction molecule 1 (STIM1), the putative calcium sensor in the ER, to enhance significantly the store-operated CRAC current (19). In addition, the defective influx of Ca2+ into T cells from SCID patients has been attributed to a point mutation in human ORAI1 (1). The replacement of wild-type ORAI1 into T cells from patients carrying the ORAI1 SCID mutation results in restored Ca2+ influx and CRAC channel current. Human ORAI1 is located on chromosome 12q24 and single nucleotide polymorphism (SNP) analysis has revealed that in SCID patients there is a missense mutation in exon 1 corresponding to a C-to-T transition at position 271 of the coding sequence that results in the replacement of wild-type Arg91 with Trp. Wild-type residue Arg91 is located within the first of the four putative transmembrane domains of Orai1, which is highly conserved between species and, thus, likely a critical residue for the function of Orai1 as a CRAC channel.
The specific role of Orai1 has not yet been fully determined, however, it is clear that the mutation in Orai1 observed in the SCID patients prevents CRAC channel function and, therefore, T cell function. How does the missense mutation observed in these SCID patients have such a dramatic effect on the functioning of the CRAC channel? The expression and location of both wild type and mutant Orai1 are similar, as demonstrated through the use of antibodies specific for extra- and intracellular regions of the mature protein (17). Therefore, receptor expression and localization are unlikely to be reason for the malfunction of Orai1 observed in the T cells of these patients.
Targeted mutation analysis of Orai1 has revealed the importance of other residues within the Orai1 protein for CRAC channel function. A Glu180→Asp180 mutation significantly alters the functioning of the CRAC channel without altering its activation kinetics. This mutation occurs within the highly conserved S1-S2 loop, located within the putative pore region of Orai1, and leads to altered ion selectivity of the CRAC channel. As a result, the channel is outwardly rectifying and specific for monovalent (i.e., Na+ or K+) rather than bivalent Ca2+ cations. In addition, a charge-neutralizing mutation at residue 180 produces an Orai1 protein that is non-conductive and acts as a dominant-negative subunit (18). These results reveal the dramatic effect a single point mutation can have on the functioning of the CRAC channel; however, just how the mutation observed in the SCID patients effects the function of Orai1 has yet to be elucidated. What is of particular interest is that the mutation at residue 180 is located in a transmembrane domain of the protein rather than in the putative pore region. It will be interesting to establish the extent of Orai1 mutations in patients with forms of SCID that are yet to be classified. Ca2+ channel activity is not routinely measured in the T cells of SCID patients consequently defects in CRAC channel function and Orai1 mutations may therefore not have been previously diagnosed.
The diagnosis, treatment, and therapy for SCID have come a long way since the first cases of SCID were reported in the 1970s. Previously, because of limited understanding of the disease and the lack of proven treatment, patients had to be kept in scrupulously isolated environments to prevent serious infection. As our understanding of SCID and other PIDs has advanced, in large part arising from the advances in molecular genetic techniques, better treatment options have emerged and patients are more able to cope with their defective immune systems.
Defects in Cytokine Receptors and in Cytokine-Mediated Signaling Leading to SCIDa
Defects in Other Lymphocyte-Specific Genes Leading to SCIDa
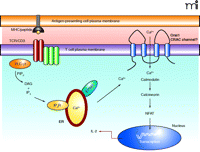
Calcium is essential for the effector processes that occur downstream of T cell receptor activation. T cells recognize antigen in the form of a peptide fragment presented in the context of MHC via the TCR/CD3 complex on the cell surface. (TCR appears as two dark red ovals; CD3 complex appears as three green ovals adjacent to TCR.) Activation results in the phosphorylation of PLC-γ1, leading to the hydrolysis of PIP2, generating DAG and IP3. IP3 causes the release of calcium from stores within the endoplasmic reticulum which is detected by STIM1. This causes the opening of CRAC channels in the plasma membrane and Ca2+ influx leading to NFAT activation, via calmodulin and calreticulin, and the transcription of genes responsible for T cell effector function (i.e., IL-2). TCR, T cell antigen receptor; MHC, major histocompatability complex; PLC-γ1; phospholipase C–γ1; PIP2, phosphatidylinositol bisphosphate; DAG, diacylglycerol; IP3, inositol-3,4,5-trisphosphate; IP3R, inositol trisphosphate receptor; ER, endoplasmic reticulum; NFAT, nuclear factor of activated T cells; CRAC, Ca2+-release-activated Ca2+channel; IL-2, interleukin-2.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Massimo Gadina, PhD, is a Senior Lecturer in the Department of Microbiology and Immunology at Queen’s University, Belfast. His research focuses on signaling pathways that control lymphocyte activation, adhesion, and migration. He is a big fan of AC Milan. Address correspondence to MG. E-mail m.g.gadina{at}qub.ac.uk; fax +44-(0)28-90325839.

Benjamin McNaull studied Life Science as undergraduate at Queen’s University in Kingston, Ontario, Canada and then continued his studies in Kingston to complete an MSc in Pharmacology. He is currently pursuing a degree in medicine and is in his third year of studies at Queen’s University Belfast. He is interested in research medicine and how it impacts patients in the clinical setting. In his spare time he enjoys running, hiking, cooking, and playing the piano.
Helen Carroll received her PhD from the University of Newcastle upon Tyne, England in the field of Transplant Immunology. She is currently working as a Postdoctoral Research Associate in the Department of Microbiology and Immunology at Queen’s University, Belfast, studying cytokine regulated genes.

