Ockham’s Razor and Selective Androgen Receptor Modulators (SARMs): Are We Overlooking the Role of 5α-Reductase?
The androgen receptor (AR) is a member of the nuclear receptor superfamily, and an important drug target. Various AR ligands (1–4) have been discovered and developed for the treatment of male hypogonadism, prostate cancer, muscle wasting, anemia, and benign prostate hyperplasia (BPH). AR ligands can be classified as agonists (androgens) or antagonists (antiandrogens), based on their pharmacological activity (i.e., ability to activate or inhibit the transcription of AR target genes); or as steroidal and nonsteroidal ligands based on structure. Endogenous androgens include testosterone and dihydrotestosterone (DHT); both are steroidal AR agonists. Testosterone is the major circulating androgen and is converted, in a tissue-specific manner, to DHT by 5α-reductase (in prostate and skin) or to estrogen by aromatase (in adipose tissue, bone, and CNS). Pharmacologically, androgen actions in reproductive tissues, including the prostate, seminal vesicle, testis, and accessory structures, are commonly referred to as androgenic effects, whereas the growth-promoting effects of androgens in muscle and bone are recognized as anabolic effects.
A variety of testosterone preparations (e.g., transdermal patches and injectable esters) and steroidal analogs (e.g., 17α-alkylated androgens and 19-norandrogens) have been developed. However, virilizing androgenic side effects, including acne and hirsuitism, concerns related to hepatotoxicity (e.g., 17α-alkylated androgens), serum lipid profiles and the incidence or severity of prostate diseases, and less than optimal pharmacokinetic characteristics (e.g., testosterone is not orally available, and has short half life in vivo) have limited their clinical application. Various nonsteroidal pharmacophores were introduced in the 1970s as a means to overcome drug design limitations imposed by the rigid steroidal plane. Nonsteroidal AR antiandrogens, including bicalutamide, flutamide, and nilutamide, remain an important and widely used treatment in the management of prostate cancer. Although these nonsteroidal anti-androgens exhibit high specificity for AR and are orally available, they do not possess meaningful tissue selectivity. Along with the blockade of AR action in the prostate, antiandrogens also block AR actions in other target tissues, including anabolic tissues (e.g., skeletal muscle and bone) and the hypothalamus-pituitary-testis axis.
The concept of a tissue-selective AR modulator (SARM) was first documented in 1999 (5). An ideal SARM is expected to have: 1) high specificity for the AR; 2) desirable oral bioavailability and pharmaco-kinetic profile; and, most importantly, 3) desirable, tissue-selective pharmacological activities. The major discriminating criterion is tissue selectivity of the ligand in vivo. With much improved tissue selectivity, these ligands should allow previously untenable therapeutic applications (6). For example, anabolic androgens could be used for the treatment of osteoporosis, muscle wasting conditions such as frailty and those caused by severe burn injury, cancer, and end-stage renal disease and AIDS. They could also be used for hormone replacement therapy in elderly men, and even in women, without concerns related to virilizing effects. On the other hand, tissue-selective antiandrogens could be advantageous for the treatment of BPH and prostate cancer, by specifically blocking androgenic actions in the prostate without abolishing needed effects on muscle, bone, or libido (3, 6).
Overall, the discovery and development of SARMs is at an early stage, with many compounds still under preclinical development and a handful now completing either phase I or phase II clinical trials. No SARM has entered the market to date (3). Most of the SARMs identified to date are nonsteroidal anabolic agents, with the first generation [aryl propionamide (7) and quinoline (8) analogs] reported in 1998. The aryl propionamide SARMs were the first to demonstrate tissue selectivity in vivo in 2003 (9), followed later that year by a tetrahydroquinoline (THQ) SARM (10), quinoline (11) SARM in 2006, and hydantoin (12) SARM in 2007. All of these anabolic SARMs demonstrate some degree of tissue selectivity in castrated animal models, with stronger agonist activities in anabolic tissues (e.g., levator ani muscle) than in androgenic tissues (e.g., prostate).
Ostrowski et al. (12) recently reported the results of their studies with a modified hydantoin SARM, BMS-564929. This SARM (Figure 1B⇓) is one of the most potent (ED50 of 0.0009 mg/kg in levator ani muscle) and highly tissue selective SARMs (160-fold, calculated as the ratio of ED50 values in the prostate and levator ani muscle) reported to date. The claimed potency of BMS-564929 in muscle may have been overstated, however, considering the fact that the report by Ostrowski et al. is the only one to suggest that castration leads to complete shrinkage of the levator ani muscle (Figure 1B⇓) (9, 10, 13). It is important to note that BMS-564929 also potently suppresses serum levels of luteinizing hormone (LH), the gonadotropin responsible for stimulating testicular production of testosterone. For example, the ED50 for BMS-564929 for LH suppression (0.008 mg/kg) was significantly lower than the doses required for 100% muscle stimulation (0.1 mg/kg), indicating that profound suppression of endogenous LH levels and testosterone production occurs within the range of doses required for anabolic activity. Selectivity with regard to gonadotropin suppression represents a significant barrier to the clinical use of SARMs. Clinical studies with BMS-564929 will undoubtedly need to address this issue in humans, particularly as related to its intended use in the treatment of andropause and age-related functional decline, where further declines in endogenous testosterone levels owing to drug treatment would likely be deemed clinically unacceptable.
Various mechanisms of action have been proposed to explain the tissue selectivity of SARMs, including differential tissue distribution of the ligands, potential interactions with 5α-reductase and/or aromatase at the tissue level, ligand-specific regulation of gene expression (genomic action), and/or nongenomic actions at molecular level. Although most of these concepts have been “adopted” from more advanced research in the area of selective estrogen receptor modulators (SERMs)––with the idea that the mechanisms of action identified for SERMs should apply to other selective nuclear receptor modulators like the SARMs––there are fundamental differences between androgen and estrogen physiology and signaling that are commonly overlooked. First of all, there are two forms of ER (ERα and ERβ) but only one endogenous ligand (estrogen). ERα and ERβ exhibit differences in their structure, ligand discrimination, tissue distribution patterns, transcriptional properties, and biological roles. The presence of two forms of ER provides a natural way to regulate estrogen action in different tissues.
In contrast, there is only one AR but two endogenous ligands (testosterone and DHT). The presence of two endogenous androgens provides a different mechanism to regulate androgen action in different tissues. 5α-Reductase converts testosterone to DHT, and is expressed in a tissue-specific manner. Type II 5α-reductase, in particular, is highly expressed in the prostate (androgenic tissue), but at relatively minor levels in skeletal muscle and bone (anabolic tissue). As such, DHT is the predominant androgen (> 95% under physiological conditions) (14) in the prostate, whereas testosterone is the major circulating androgen and functional form in muscle and bone. Because DHT is a more potent androgen in the prostate, testosterone action is “amplified” in the prostate, thereby abolishing its “tissue-selectivity” (Figure 1A⇓). Furthermore, as shown by Wright et al. (14), 5α-reductase inhibition by finasteride elevates testosterone to the dominant androgen in the prostate (>80%), showing much lower potency than DHT in stimulating prostate growth, and causing a significant right-shift of the dose response curve (Figure 1A⇓).
Has the important role of 5α-reductase in AR and SARM been overlooked? Ostrowski et al. (12) presented data regarding the interaction between BMS-564929 and aromatase, but not 5α-reduc-tase. Although the conversion of testosterone to estrogen does play some role in the development of prostate disease, its contribution to the overall growth of the normal prostate is rather limited (15). Considering BMS-564929 is a structural derivative of nilutamide, a nonsteroidal antiandrogen that does not interact with 5α-reductase (16), it is not likely that BMS-564929 would be a 5α-reductase substrate or inhibitor. As the only active androgen in the prostate, the dose-response curve of BMS-564929 in the prostate demonstrated a significant right shift (Fig 1B⇓) similar to that observed with testosterone alone (with the presence of finasteride, Fig 1A⇓). Likewise, in vitro studies of BMS-564929 action in rat prostate epithelial cells that express 5α-reductase in the report by Ostrowski et al. [see Figure 3 in (12)] also demonstrate that the actions of testosterone are amplified, whereas those of the SARM are not, resulting in a right-shift of the dose-response profile for the latter.
Although more intriguing explanations suggestive of novel AR conformations and tissue-specific cofactor recruitment are often sought, existing evidence with BMS-564929 and other SARMs strongly suggests that tissue-specific expression of 5α-reductase plays an overwhelming, if not complete, role in determining the tissue selectivity of SARMs. Recent studies (17) with a nonsteroidal AR ligand indicate that differential tissue distribution is unlikely to contribute to differences in the pharmacologic responses observed for nonsteroidal AR ligands in the prostate and muscle. Similarly, although changes in AR function are initiated by ligand-induced conformational changes of the ligand binding domain (LBD), crystal structures of SARM-bound AR LBD (aryl propionamide and hydantoin SARMs) did not reveal conformational changes as significant as those observed in SERM-bound ER LBD (12, 18), particularly the AF2 region (19, 20). On the other hand, testosterone- and DHT-bound AR LBD structures were considered “essentially identical” (12) despite the considerable differences in their androgenic actions in the prostate (Figure 1A⇓). Although crystal structures are very useful for determining ligand binding mechanisms, it is difficult to predict potential changes in receptor function that would occur under native conditions. Subtle changes in protein conformation, surface topology, or both that might affect protein-protein interaction, and receptor function needs to be assessed more carefully under native conditions to identify meaningful consequences in function, if any. Furthermore, although peptide mapping and protein-protein interaction assays (21, 22) have identified ligand-specific interaction profiles of the receptor, the significance of these differential profiles in function has not been fully evaluated at tissue level or in vivo. As such, it is unclear if these differences play any significant role in ligand-stimulated AR action in different tissues. Therefore, much more work needs to be done in order to prove that certain molecular mechanisms contribute equally or at all to the tissue selectivity of SARMs.
In conclusion, there has been considerable progress in the discovery and development of SARMs in the past decade. SARMs with much improved specificity for AR, in vivo pharmacokinetic profiles, and higher degree of tissue selectivity will undoubtedly expand the clinical applications of androgens. Unlike SERMs, the tissue selectivity of SARMs may be more related to the tissue-specific expression of 5α-reductase and lack of interaction between SARM and 5α-reductase. Further studies are required to determine if the molecular mechanisms identified in SERM research can also be applied to SARMs. Certainly, ligand-specific conformation changes and tissue-specific differences in cofactor expression and interaction play a role in SARM pharmacology; the question remains: to what extent do they contribute to the observed differences in tissue response (e.g., prostate and muscle) during SARM treatment? Paraphrasing the fourteenth century philosophy of William of Ockham, “all things being equal, the simplest solution tends to be the best one.” Although 5α-reductase expression and activity may not explain all of the tissue-selective pharmacologic actions of SARMs, it may indeed play the predominant but less beguiling role in the observed tissue-selective pharmacologic activity of SARMs.
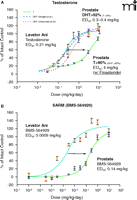
Tissue-specific expression of 5α-reductase contributes to the tissue selectivity of SARMs. (A) Partial agonist activity of testosterone in the prostate was revealed when co-administered with type 2 5α-reductase inhibitor finasteride. Dose-response curves of T in the levator ani muscle (•) and T in the prostate (○ and dotted line) were redrawn with permission from Ostrowski et al. (12); and dose-response curves of T in the prostate with (▴) or without (▵ and dash line) co-administration of finasteride were redrawn with permission from Wright et al. (14). T data from two different studies [Wright et al. (14) dotted line; and Ostrowski et al. (12) dash line] are compared directly here, considering the fact that both studies used a re-growth model (T treatment after castration caused complete shrinkage of the prostate) with SD rats of similar age and subcutaneous administration of T. Furthermore, the dose-response curves of T in the prostate (largely DHT due to 5α-reductase action) obtained from two studies largely overlap, with similar ED50 values between 0.3~0.4 mg/kg/day, showing that these two studies are comparable. (B) Partial agonist activity of SARM (hydantoin analog BMS-564929) in the prostate when administered alone. BMS-564929 was orally administered because it is orally available. Dose-response curves redrawn with permission from (12).] T, testosterone.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Wenqing Gao, PhD, is an Assistant Professor in the Department of Pharmaceutical Sciences, at the University at Buffalo, SUNY. She received her Ph.D. from The Ohio State University where she studied the pharmacology of selective androgen receptor modulators (SARMs) in Dr. James Dalton’s laboratory. Her current research interests include structural biology and molecular mechanism of actions of nuclear receptors and coregulators.

James T. Dalton, PhD, is a Professor of Pharmaceutics in the College of Pharmacy at The Ohio State University. He is currently on leave of absence from the university and serving as Vice-President of Preclinical Research and Development at GTx, Inc., Memphis, TN (www.gtxinc.com), a men’s health biotech company leading clinical development of nonsteroidal selective androgen receptor modulators. Dr. Dalton is a co-inventor on over fifty US and international patents and patent applications on SARMs. His research interests include the molecular, preclinical, and clinical pharmacology of novel drugs, with an emphasis on selective nuclear receptor modulators and anticancer agents. E-mail: DALTON.1{at}OSU.EDU, fax 614-292-7766

