Cyclic Nucleotide Signaling Mechanisms in Trypanosomes: Possible Targets for Therapeutic Agents
Abstract
Trypanosome infections cause several major human diseases, including sleeping sickness and Chagas disease, which affect millions of people in Africa and South America, respectively. Although adenosine 3′,5′-monophosphate (cAMP) signaling and regulation have been widely studied in mammalian systems, and these pathways provide targets for the treatment of numerous pathologies, a molecular understanding of cAMP signaling in trypanosomes remains incomplete. Recent studies in these parasites, however, have revealed diverse families of adenylyl cyclase and phosphodiesterase that regulate cAMP concentrations. Importantly, these enzymes differ pharmacologically and biochemically from their mammalian counterparts. In this review, we discuss recent developments, emerging ideas, and gaps in knowledge in this area of research, highlighting aspects of enzymes in the cAMP signaling pathway that may be good targets for antitrypanosomal drug therapy.

Introduction
Trypanosomes are unicellular protozoans that live as parasites of insects, birds, fish, and mammals. All of these parasites are flagellated and contain a unique DNA structure called the kinetoplast, which is a concatenated network of circular DNA in the mitochondrion. Two trypanosome species, Trypanosoma brucei and T. cruzi, are the cause of important human diseases. T. brucei, endemic to sub-Saharan Africa, causes African trypanosomiasis, or sleeping sickness. This is a severe disease that causes between 50,000–200,000 deaths annually (1). The parasite is transmitted to humans or livestock by the tsetse fly, and infection is initially characterized by malaise and irregular fevers. When left untreated, the parasite invades the central nervous system, leading to coma and death. The South American trypanosome, T. cruzi, causes Chagas disease in South and Central America (2). This disease is transmitted to humans primarily by various biting reduviid bugs of the Triatominae subfamily (including Assassin bugs, Cone-nosed bugs, and Kissing bugs), by transfusion of infected blood, or by transmission to newborn children from infected mothers. Patients infected with T. cruzi typically present with symptoms of fever, swollen lymph nodes, and swollen, infected bite sites termed chagomas, but often remain without symptoms for years. During this time the parasites invade internal organs of the body, causing severe damage to the heart, intestine, and esophagus, resulting in progressive weakness. It is estimated that there are over fifteen million individuals worldwide infected by T. cruzi, with millions more at risk (3). Another closely related group of parasites belonging to the Leishmania genus cause various forms of leishmaniases across the tropical and temperate world. These diseases exhibit diverse clinical symptoms and collectively infect over two million people annually.
Life Cycles of Trypanosoma Bruceiand T. Cruzi
The life cycles of trypanosomes are quite complex, owing to their survival in insect and mammalian hosts. T. brucei exist in the mammalian bloodstream as rapidly proliferating morphologically slender forms. As parasite numbers increase, many of these slender forms transform into a cell-cycle arrested morphological form (“short stumpy”) that does not proliferate in the mammalian host but is adapted to survive in the tsetse fly. When taken up by the tsetse fly, the parasites transform into procyclic forms, which proliferate in the fly mid-gut, subsequently migrate to the tsetse salivary gland, and remain as epimastigote forms (Figure 1⇓).
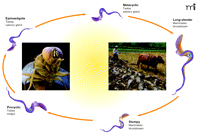
The life cycle ofT. brucei. The trypanosome T. brucei, the causative agent of Afrtican sleeping sickness is capable of living in an arthropod vector (tsetse fly) and in humans. Thus it is no wonder the parasite has a very complex life cycle allowing it to survive (and thrive) in its hosts. Trypanosomes drawn for illustrative purposes only and lack particular details specific to each stage.
Epimastigote forms finally become non-proliferative meta-cyclic forms, which are primed for survival in the mammalian bloodstream (4). Similarly, T. cruzi has a complex life cycle in both insect as well as mammalian host, surviving as amastigote, sphaeromastigote, epimastigote, and finally metacyclic forms within the reduviid insect. The metacyclic form parasitizes mammalian hosts, where the trypanosome survives as intermediate, slender and broad trypanosome forms (5).
Current Drug Therapy
At present, there are limited choices for the treatment of African sleeping sickness (6, 7) or Chagas disease (7–9). Two drugs are in common use for the treatment of first stage African sleeping sickness: pentamidine, to treat sleeping sickness caused by T. b. gambiense; and suramin, to treat T. b. rhodesiense. Both drugs are over fifty years old and have numerous undesirable side effects on the heart, blood pressure, and blood sugar, as well as other lesser side effects. For late-stage sleeping sickness, only two somewhat effective drugs are available: melarsoprol, an arsenic derivative often associated with severe arsenic toxicity at effective doses, and eflornithine, which is effective only against one of the major sleeping sickness–causing T. brucei subspecies, T. b. gambiense. In addition to being incompletely effective and toxic, these drugs have strict dosing regimens, for which full patient compliance is extremely difficult (6, 7, 10).
Similarly, only two drugs are widely available for the treatment of the acute stage of Chagas disease: nifurtimox and benznidazole. Both drugs have limited effectiveness in the late stage, and also show severe side effects ranging from anorexia and nausea to cutaneous eruptions, generalized edema, and depression of bone marrow production (7–9). Overall, there is an urgent need to identify new targets and new agents for anti-trypanosomal therapy.
Adenosine 3′,5′-Monophosphate (cAMP) Signaling in Trypanosomes
The second messenger molecule adenosine 3′,5′-monophosphate (cAMP) is one of the most widely studied signaling molecules in mammalian and other eukaryotic systems (11), with important roles in cell differentiation and growth, hormone regulation, sensory signal transduction, and various diseases. In most eukaryotes, cAMP is synthesized by adenylyl cyclases (ACs), degraded by phosphodiesterases (PDEs), and acts through effector proteins such as protein kinase A, exchange proteins directly activated by cAMP (EPACs), and ion channels (11). These pathways have proven to be important clinically, and numerous drugs target receptors coupled to ACs or PDEs (12, 13). Given the success of drugs targeting the cAMP pathway in humans, it would seem that the cAMP signaling pathway in trypanosomes could provide an attractive target for the development of new therapeutics. To date, however, this has not happened because of a poor understanding of cAMP signaling pathways in trypanosomes.
A primary role suggested for cAMP in both T. brucei and T. cruzi is to regulate various differentiation pathways, although evidence for such a role is incomplete, owing to the complex life cycles of trypanosomes. Early studies reported different amounts of cAMP in reproducing and in differentiated T. lewisi (14), as well as alterations in cAMP concentrations during developmental cycles of T. brucei (15). Additionally, an increase in cAMP was observed in T. cruzi during differentiation of the insect stage epimastigote to the infectious metacyclic trypomastigote (16). These results have been interpreted to suggest that cAMP may be an important regulator of these processes.
The mechanisms causing these changes in cAMP are not fully understood. In T. brucei, increased AC activity has been observed during transformation from mammalian bloodstream to procyclic (insect stage) forms. An initial peak of AC activity coincides with the release of the variable surface glycoprotein (VSG) coat, and a second peak occurs when the cells complete the first division, but before proliferation. VSG release, however, is not affected by the increased cyclase activity (17, 18), and in mutant trypanosomes lacking VSG lipase (the enzyme that cleaves the GPI anchor from the VSG polypeptide chain and releases the protein), AC activity increases in the absence of VSG release and vice versa (18). This study clearly demonstrated that whereas VSG release and activation of AC activity can occur under the same set of stimuli, they are not obligatorily coupled (18).
Unfortunately, many early studies reported variable and sometimes conflicting results. For example, one group of investigators working with T. cruzi used cAMP analogs or activators of mammalian ACs (such as cholera toxin) to induce the transformation of T. cruzi epimastigotes to metacyclic forms (19). In another study, common mammalian PDE inhibitors (e.g., theophylline) did not affect T. brucei proliferation or intratrypanosomal cAMP (20). Other studies showed that high concentrations of dibutyryl cAMP [a cell-permeable cAMP analog that activates cAMP-dependent protein kinase (protein kinase A, or PKA)] had no effect on differentiation in vitro (21). In other apparently conflicting studies, cell-permeable cAMP analogs (or a putative trypanosome PDE inhibitor, etazolate) transformed slender, proliferating bloodstream forms of T. brucei into cell-cycle arrested, stumpy-like bloodstream forms and this effect was attributed to cAMP (22, 23). In T. cruzi, analogous studies have led to the suggestion that cAMP can decrease proliferation by reducing DNA or RNA synthesis (24). Many of these studies have assumed, often without good direct experimental evidence, that agents known to work in mammalian systems will also have similar activities in trypanosomes. These and other suggested roles of cAMP in T. brucei or T. cruzi have also been reviewed in more detail elsewhere (25, 26).
Established roles for cAMP in trypanosomes also remain unclear because of our limited understanding of the molecular interactions of cAMP-influenced effector proteins. Recent work has identified and characterized several unique families of ACs and PDEs, both of which appear to differ biochemically and pharmacologically from their mammalian counterparts. The complete sequences of the T. cruzi and T. brucei genomes may provide new knowledge of the targets for cAMP signaling. Indeed, some progress has been made in identifying and characterizing putative cAMP effector proteins. The rest of this review summarizes recent knowledge of ACs and PDEs in T. brucei and T. cruzi, identifies putative cAMP effector proteins in the trypanosome genomes, and points to developments that identify possible new roles for cAMP in trypanosomes.
Trypanosome Adenylyl Cyclases (ACs)
Trypanosome AC activity was partially purified from T. cruzi membranes nearly thirty years ago (27). Eventually, a complex multigene family encoding several ACs was described in T. cruzi (28). These studies also reported that AC genes were present on at least six different chromosomes. Several AC genes also have been described for T. brucei (29, 30). Early studies in T. brucei showed that the polycistronic transcription unit of the variant surface glycoprotein (VSG) gene encoded an AC gene as well (31, 32). In T. brucei, some ACs are transiently expressed in the mammalian bloodstream forms, with each VSG expression site containing a different AC gene (e.g., expression site associated gene 4, or ESAG4). Others are constitutively expressed across life-cycle stages and are called “genes related to ESAG4” (GRESAG4) isozymes. As discussed earlier, a peak in cyclase activity is coincident with (though not causal for) VSG release. The specific roles of these ESAG4 cyclases and their interplay (if any) with VSG expression remain undetermined but are intriguing.
Unlike the G protein–coupled, multiple transmembrane domain mammalian Class I ACs (33), trypanosome cyclases belong to the Class II cyclases, found exclusively in protozoa. All trypanosome cyclases have long, diverse extracellular regions, a single transmembrane domain, followed by a single, well-conserved intracellular catalytic region. The catalytic domains of trypanosome cyclases exhibit a high degree of sequence similarity to both the mammalian adenylyl and guanylyl cyclases (GCs) and are therefore easily identified by bioinformatics approaches as typical cyclase catalytic domains. For example, all contain the arginine and asparagine residues thought to be required for catalysis (33).
A recurring problem in the interpretation of many studies of cAMP signaling in trypanosomes relates to the methods used to test for such signaling. For example, in studies of cAMP synthesis, it is often assumed that the structures and regulation will be essentially the same in trypanosomes as it is in mammals. Similarly, it is often assumed that the same drugs and probes that work in mammals will work similarly in trypanosomes. Unfortunately, this often is not likely to be true. The structures of the trypanosome and mammalian ACs differ substantially in their catalytic domains, as the trypanosome cyclases lack a binding region for heterotrimeric G proteins (which bind and regulate mammalian cyclases) (34), and do not require G proteins for biochemical activity (35). Additionally, heterotrimeric G proteins cannot be found in the now complete trypanosome genomes. Finally, mammalian cyclase agonists, such as forskolin or cholera toxin, do not appear to activate trypanosome ACs (18, 25, 34).
More recently our understanding at the molecular level of trypanosome ACs has been greatly improved by the recently completed genomes of both T. brucei and T. cruzi (36, 37). These genome sequences reveal at least fifty putative AC genes scattered across several different chromosomes in T. brucei. It is possible that some of these sequences may be “pseudogenes,” or if expressed, may not have catalytic activity (a phenomenon observed with many trypanosome genes). Although a complete annotation of the genome will be required to reveal the number of true cyclases, the number of AC genes in T. brucei is likely to be very large. Similarly, the T. cruzi genome appears to have at least eighteen predicted AC genes, but less is known about their expression patterns or specific function; however, the catalytic domains of ACs of T. cruzi share much structural similarity with those of T. brucei (28). This multiplicity of genes may make it difficult to find effective drugs that target all of the trypanosome cyclases without affecting mammalian enzymes.
Biochemical and structural studies have now addressed some aspects of T. brucei cyclase activation as well as catalysis. Although not G protein–coupled, the trypanosome cyclases appear to require dimerization for activity. In a study utilizing recombinant, purified GRESAG4.4 AC catalytic domains, the expressed catalytic domains spontaneously dimerized but showed low basal activity; nonetheless, forced dimerization with a leucine-zipper motif engineered onto the catalytic domain resulted in a large increase in cyclase activity (35). This suggested that conformation changes induced by the leucine zipper–mediated dimerization mimicked a native, activated form of this T. brucei cyclase and that specific ligand-induced conformational changes could activate T. brucei AC catalytic subunit dimers. Interestingly, older work has shown that a region adjacent to the AC gene expresses a sequence encoding a leucine-rich repeat domain, similar to the leucine-rich regulatory domain of yeast ACs (32), perhaps hinting at possible physiological regulation. Subsequent work, however, has shown that this leucine-rich protein accumulates in the nucleolus, interacts with a Pumilio protein family member, and appears to regulate RNA stability in trypanosomes. This protein does not appear to be involved in cAMP signaling (38, 39).
The molecular mechanism for ATP conversion to cAMP by the catalytic domains of the trypanosome cyclases has been addressed experimentally using biochemical and X-ray crystallographic approaches for two of the GRESAG4 AC catalytic domains of T. brucei (34, 35). One question of particular interest is what makes the cyclases insensitive to mammalian cyclase activators or inhibitors, while retaining the core-catalytic residues involved in ATP-to-cAMP conversion? This question has been partially answered by solution of the crystal structures of the two GRESAG4 AC isoforms (34). Although the structures showed a high degree of overall similarity to mammalian AC C1 and C2 domains, they revealed some unique features absent in mammalian cyclases, including a large insertion of approximately thrity-six amino acids between the third α-helix and fourth β-sheet, and also revealed a highly ordered C terminus that forms a cleft between the helical domain and a long loop (34).
In spite of large similarities in the nucleotide binding sites of the T. brucei cyclases and mammalian cyclases, the T. brucei cyclases show substantial differences in their responses to the so-called P-site mammalian cyclase inhibitors (i.e., 2′-deoxyadenosine or its derivatives) (40, 41). The reasons for this were not fully apparent from the structures of the T. brucei cyclase isoforms. Finally, the structures of the two cyclases also revealed a small internal cavity and a unique trypanosomal insertion (called the Δ subdomain) in a region where mammalian cyclases bind regulatory ligands (34). The authors speculated that this region might bind ligands that affect T. brucei cyclase dimerization and activity. A model of a T. brucei AC dimer (of GRESAG3.1) reveals the extracellular region, a single transmembrane domain, and an intracellular catalytic domain (Figure 2⇓).
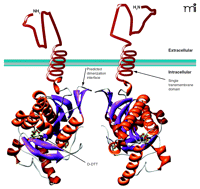
Representation ofT. bruceiadenylyl cyclases (ACs). The adenylyl cyclases (ACs) found in T. brucei contain a variable extracellular domain, a single transmembrane domain, and a catalytic domain. The catalytic domain shown is that of the GRESAG4.1, which can dimerize, leading to the activation of these enzymes. Development of synthetic ligands that force the dimerization of these ACs would provide valuable research tools to study the role of these enzymes in trypanosomes and may have possible therapeutic uses. Additionally, the crystal structure shows a molecule of (2S,3S)-1,4-dimercapto-2,3-butanediol (D-DTT) bound at a site within the catalytic domain. This site might be targeted through the development of agonists or antagonists designed to bind the AC and modulate its activity.
Pharmacologically, it would seem possible to design agents (similar to the engineered leucine zipper desribed above) that would forcibly dimerize trypanosome cyclases or to design small molecules to fit into the putative regulatory internal cavity, enabling activation or inhibition of these enzymes. These agents could be tremendously powerful tools in analyzing the roles of ACs and cAMP in trypanosomes and could prove therapeutically useful.
Natural ligands for the T. cruzi or T. brucei cyclases still remain unknown. Trypanosomes might respond to natural regulatory ligands that bind to the extracellular domain or cytosolic domain of trypanosome cyclases. To date, no such putative regulatory molecules have been definitively identified. A peptide from the hindgut of Triatoma hematophagous, an insect vector of T. cruzi, was suggested to stimulate T. cruzi cyclase activity and appeared to promote differentiation from epimastigote to metacyclic insect life-cycle forms (42). In another report, proventricular and esophageal tissue from the tsetse fly appeared to stimulate AC activity in T. brucei (43). These observations are particularly intriguing because they suggest that trypanosome differentiation may be triggered by factors synthesized within the host and not in the parasite itself; however, these studies have not been independently verified
Finally, several trypanosome ACs are known to respond to Ca2+ or calmodulin. A calcium-activated cyclase has been identified and characterized in T. cruzi that interacts with the paraflagellar rod (PFR) protein (44). Furthermore, calcium appears to stimulate AC activity from a recombinant T. brucei ESAG4, and calcium stimulation of cyclase activity also has been observed in the bloodstream form of T. brucei (45, 46). However, endogenous ligands that directly or indirectly regulate the calcium-dependent trypanosome ACs have yet to be identified, making the study of physiological roles for these numerous cyclases more challenging.
Despite the advances in identifying and characterizing trypanosome ACs, unique and specific roles for these numerous ACs in cAMP signaling and trypanosome biology have not been elucidated. Some of the trypanosome ACs appear to be localized in the flagellar region or interact with flagellar proteins (in both T. cruzi and T. brucei) (31, 44, 47), but such localization can only hint at possible AC functions. Are all the expressed trypanosomal cyclases required for cAMP production? Or do they perhaps have other functions? Given their diverse extracellular regions, each AC might act as a sensor for extracellular ligands or stimuli, or could be part of a large scaffolding complex that binds other proteins with unique functions. It is noteworthy that no GC has been identified in trypanosomes, nor has any role for guanosine 3′,5′-monophosphate (cGMP) been proposed in these parasites; nevertheless, given the similarity of these ACs to mammalian GCs in primary sequence identity as well as the overall domain organization of the enzymes, it may be worth asking whether any of these putative ACs can also synthesize cGMP. These and many other questions about trypanosome ACs remain unanswered.
Phosphodiesterases in Trypanosomes
cAMP-specific PDEs terminate cAMP signals by breaking down cAMP to 5′-AMP. The earliest demonstration of PDE activity in bloodstream from T. brucei gambiense lysates occurred more than thirty years ago (48). Many early (and some recent) studies have used mammalian PDE inhibitors, such as methylxanthines [including 3-isobutyl-1-methylxanthine or (IBMX), caffeine, or theophylline], to inhibit trypanosomal PDEs, and speculated that certain effects arose from PDE inhibition (and, therefore, were cAMP-dependent). Confounding the issue, other early work showed that the PDE activity in T. cruzi was not inhibited by the concentrations of methylxanthines that are effective with mammalian PDEs (49). Much of our current molecular, biochemical, and pharmacological understanding of PDEs in trypanosomes comes from cloning and characterization studies of PDEs from both T. cruzi and T. brucei.
Four Families of Phosphodiesterases (PDEs) inT. CruziandT. Brucei
The recently completed trypanosome genome databases have provided substantial information about the organization of PDEs in these parasites. These genomes reveal four well-conserved families in both T. brucei and T. cruzi (as well as L. major) and have allowed for a unified nomenclature to be proposed (50). The four PDE families found in trypanosomes, TxxPDEA–D (where “xx” represents “br” for T. brucei or “cr” for T. cruzi), are illustrated in the phylogenetic tree (Figure 3⇓). All four PDEs belong to the Class I group of PDEs, similar to the large number of Class I PDEs found in mammals (12).
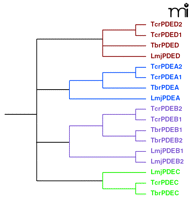
A phylogenetic tree depicting the four different phosphodies-terase (PDE) families in trypanosomes andL. major. The four phosphodi-esterase (PDE) families are conserved across trypanosome and leishmania species. Sequence differences—particularly in the regulatory domains—can be substantial not only across genera but also within species. Thus, development of “broad spectrum” regulatory domain inhibitors might prove fruitless. On the other hand, catalytic site inhibitors designed against one PDE family are more likely to be effective across species and genera. Tbr, T. brucei; Tcr, T. cruzi; Lmj, L. major. The nucleotide sequences were aligned using CLUSTALW and the phylogenetic tree was built using the PHYLIP program.
The first TxxPDE family was cloned and identified in T. brucei (51–53) and constitutes the TbrPDEB family (originally called the TbPDE2 family). Recently, orthologs of the TbrPDEB family in T. cruzi (TcrPDEBs) have been identified and characterized (54, 55). The PDEB families of both species are cAMP-specific and insensitive to IBMX and other methylxanthines, as well as most other mammalian PDE inhibitors. A second PDE family in trypanosomes, the FYVE-domain containing PDEC family, was identified and characterized in T. cruzi (56). PDEC is the only trypanosome PDE identified to date capable of hydrolyzing cGMP, although it prefers cAMP as a substrate (cAMP Km ~30 μM and cGMP Km ~80 μM) and is somewhat sensitive to IBMX. Additionally, PDEC is unusual because its catalytic domain is present in the middle of the amino-acid sequence of the protein, whereas most eukaryotic PDEs have unique N-terminal regulatory domains and the catalytic domain is located near their C terminus. The PDEA family from T. brucei and T. cruzi (57, 58) possesses an unusually high Km (between 200–600 μM) for cAMP hydrolysis. It is unknown whether intracellular cAMP exists in sufficiently high concentrations in trypanosomes for this enzyme to effectively regulate cAMP. Finally, the PDED family from T. brucei and T. cruzi has a large N-terminal region of unknown function. All the PDEs mentioned show specificity or high preference for cAMP as a substrate. The only mammalian PDE inhibitor that shows a modest ability to inhibit all these enzymes is dipyridamole (IC50 ~10–50 μM) (51, 53, 56, 57). The fact that none of these trypanosome PDEs show any significant inhibition by nearly all known mammalian PDE inhibitors suggests that the active site in the trypanosome PDEs are substantially different than those of the mammalian PDEs. Therefore, it should be possible to design inhibitors that are selective for the trypanosome PDEs and not cause troublesome side effects owing to actions on mammalian PDEs. A crystal structure has not yet been reported for any trypanosomal PDE; hence, the molecular differences between mammalian and trypanosomal PDEs active sites remain to be identified.
Regulatory Domains in Trypanosomal PDEs
Specific roles for the regulatory domains of the trypanosome PDEs and their specific ligands are only recently beginning to be identified. The PDEB family of PDEs contains two N-terminal cGMP-AC-FhlA (GAF) domains, which are known to mediate allosteric cyclic nucleotide binding and dimerization in mammalian PDEs. The GAF domain of TbrPDEB2 and the GAF domains from theTcrPDEBs preferentially and co-operatively bind cAMP, in contrast to most mammalian GAF domain–containing PDEs, which preferentially bind cGMP (55, 59). Binding of cAMP to the trypanosomal GAF domains appears to regulate PDEB enzymatic activity (59). In fact, high affinity cAMP binding to a PDE GAF domain was first demonstrated in T. brucei (59), and has since been observed with mammalian PDE10 (60); however, ligands of all of the other trypanosome PDE families remain unknown. The only other domain with a known function in any of these trypanosome PDEs is the FYVE domain, a zinc-finger-like domain involved in membrane trafficking and signaling and having a high specificity for binding phosphoti-dylinositol-3-phosphate (PI3P) (61, 62). Whether the FVYE domain in the PDEC family has similar functions is not yet known.
Although some of the molecular mechanisms of cAMP action in trypanosomes are based on incomplete evidence, one vital function for cAMP in T. brucei is strongly suggested from induced RNAi-mediated knock-down of TbrPDEBs. Diminished expression of TbrPDEBs blocked proliferation of bloodstream forms of the parasite (52) without affecting procyclic (insect) forms. This knock-down also caused a dramatic alteration of phenotype, resulting in oddly shaped, multinucleate cells (52). Subsequent studies utilizing more stringently regulated plasmid vectors have found similar results (63). TbrPDEB1 and B2 have distinct localizations within the trypanosome: TbrPDEB1 is expressed exclusively in the flagella and TcrPDEB2 is distributed between the flagella and the cytoplasm (61). Additionally, when the expression of each PDE was knocked-down individually, there was no adverse effect on the bloodstream form T. brucei, suggesting that each enzyme can compensate for the other. These studies collectively suggest that cell division in double knock-down mutants was normal, but cytokinesis was severely affected by lack of both PDEs. Importantly, the authors were able to modulate the activity in the intact animal using a modified RNAi technique with good success (Box 1) (63).
RNAi-Mediated Inhibition of TbrPDEs Reduces Parasite Load In Vivo
Oberholzer et al. infected mice with trypanosomes carrying an inducible RNAi construct to knockdown (i.e., diminish) the expression of TbrPDEBs (61). Upon induction of RNAi (with tetracycline in the diet), a nearly complete elimination of infection was observed. In effect, the mice were cured. These studies not only suggested an important role for cAMP in bloodstream form T. brucei but also strongly implicate the TbrPDEB family as a good potential target for therapeutic intervention (61).
Given the success of identifying selective inhibitors for mammalian PDEs (60), finding new small-molecule inhibitors selective for the TbrPDEBs appears quite feasible. As described earlier, the TbrPDEB family of T. brucei PDEs is highly resistant to mammalian PDE inhibitors; therefore, specific trypanosome PDEB inhibitors will likely not affect human PDEs, making PDEB family members all the more attractive as therapeutic targets. Secondly, cAMP appears to be a selective ligand for the GAF domain of TbrPDEB2, and the binding of cAMP appears to regulate its PDE activity (59). Thus, it may also be possible to find new inhibitors or activators of these PDEs that act through the cAMP-binding GAF domain. Together, such pharmacological agents could be used to dissect the role(s) of cAMP in trypanosomes, and provide potential new and possibly highly effective therapeutics.
cAMP Effectors: The Missing Link
In most mammalian systems, cAMP regulates numerous physiological processes through PKA. By binding the regulatory subunit of the enzyme and releasing it from the catalytic subunit, cAMP stimulates PKA activity; however, little is known about this most prominent effector protein of cAMP in trypanosomes. A putative regulatory subunit of PKA was identified in T. brucei that immunoprecipitated with an associated kinase activity (64), but cGMP and not cAMP stimulated this kinase activity (64). This was surprising, as a GC has not been identified in trypanosomes, nor is there any measurable cGMP-dependent PDE activity in trypanosome cell lysates. The lack of cAMP stimulation of kinase activity for this putative PKA could be real if, for example, other required cofactors for cAMP stimulation of kinase activity or consensus phosphorylation sequences that differ from mammalian PKA substrates are required. The presence of a cAMP-dependent protein kinase activity was also suggested in T. cruzi (65). In this species, a catalytic kinase subunit was identified (66), cloned (67), and tentatively classified as a PKA based on sequence similarity with the catalytic domain of mammalian PKA. The expression of this putative PKA catalytic subunit also appears to be developmentally regulated (67). Similar efforts have identified putative PKA catalytic subunits in the closely related Leishmania major and L. donovani (68, 69).
The identification of a cAMP-regulated protein kinase regulatory subunit in T. cruzi that associates with the PKA catalytic subunit appears to be an important breakthrough (70). This complex showed a cAMP-dependent increase in kinase activity and was inhibited by the PKA specific inhibitor PKI (70). In trypomastigotes (and not in other T. cruzi life-cycle stages), this PKA regulatory subunit also localized to the flagellar region. This is the first clear demonstration of a PKA regulatory-catalytic subunit complex that is activated by cAMP in trypanosomes; future studies of this PKA and other putative trypanosomal PKAs will aid our understanding of cAMP signaling in these parasites. Other than the GAF domain PDEB family (55, 59) and this PKA, no other cAMP binding proteins have been definitively characterized in trypanosomes.
The recently completed genomes of T. brucei, T. cruzi, and L. major (36, 37, 71) provides valuable information towards identifying all other cAMP target proteins. T. brucei and T. cruzi genomes do not appear to have any EPAC-like guanine-nucleotide exchange factors. Analyses of the T. brucei genome to find sequences similar to the cAMP-binding domain of mammalian or yeast PKAs has revealed at least four predicted cAMP binding proteins. One of these (GeneDB gene ID number Tb11.02.2210) appears to be the characterized protein kinase A regulatory subunit (68), whereas Tb927.3.5020 (on chromosome 3) and Tb927.8.2130 (found on chromosome 8) contain predicted cNMP binding domains. Tb10.70.1860 is a particularly interesting large predicted protein found on Chromosome 10, with a predicted molecular mass of 188.2 kDa. This protein contains two tandem putative cNMP binding domains, as well as two tandem C2 domains, which are Ca2+-dependent membrane targeting domains present in phospholipases, protein kinase C, and other proteins. C2 domains bind phospholipids, inositol phosphates, and intracellular proteins (72). There are no predicted catalytic domains on this large protein, suggesting the intriguing possibility that protein is a cAMP-responsive scaffolding protein that brings signaling components together [perhaps analogous to mammalian PKA-anchoring proteins (AKAPs) that bring cAMP signaling proteins together (73)]. This is particularly interesting because the trypanosomal FYVE domain–containing PDEC family may also be involved in inositol phosphate signaling. The domain architectures of these proteins are illustrated schematically in Figure 4⇓. These four bioinformatics “hits” are likely to be an underestimation of putative cAMP-binding proteins in T. bruceibecause other proteins (such as Tb11.01.7370 and Tb11.18.0012) might contain cAMP-binding domains.
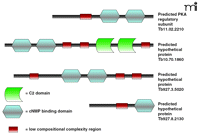
The domain organization of predicted cAMP-binding proteins in theT. bruceigenome. Some of the proteins appear less complex, containing only a single putative cAMP-binding domain, whereas others contain more than one cAMP-binding domain. The details of possible cNMP binding to these domains remain unknown. Other proteins containing predicted cNMP-binding domains are more complex, integrating multiple domains such as the C2 domain. It is hypothesized that these proteins act as scaffolding proteins bringing together diverse cAMP-mediated signaling proteins or interact specifically with other proteins and allow their regulation under the presence of specific cAMP signals.
Similarly, the T. cruzi genome has numerous predicted cAMP binding proteins (Gene IDs Tc00.1047053506227.150, Tc00.1047053504013.60, Tc00.1047053510879.50, Tc00.1047053507035.110, Tc00.1047053508523.80) that still remain unverified or characterized. Whether all of these predicted cAMP binding/regulated proteins do indeed bind cAMP is not known nor are their functions clear, but this information provides interesting directions for future investigation, and a complete annotation of both genomes will undoubtedly reveal a more precise number of cAMP effector proteins. Additionally, the T. cruzi, T. brucei, and L. major “kinomes” (i.e., the complete profile of protein, carbohydrate, and lipid kinases expressed in an organism) appear to contain three PKA catalytic subunits (74). Whether all three of these catalytic subunits associate with or are regulated by cAMP-dependent binding protein subunits is unknown but the characterization of these putative cAMP effector proteins will be crucial to understanding cAMP signaling in trypanosomes.
Roles for cAMP in Trypanosomes: Revisiting Old and Identifying New
cAMP participates in several important processes in trypanosomes, but the identification of each specific role is ongoing. The knockdown studies of the TbrPDEB family showed these PDEs to be essential in the infectious bloodstream form of T. brucei and also caused a substantial increase in cAMP concentrations, suggesting the requirement of finely regulated cAMP amounts in the bloodstream form of the parasite.
Another possible role for cAMP may involve the regulation of osmotic fluxes. Trypanosomes experience dramatic changes in osmolarity, as they pass through their diverse life-cycle stages. The efflux of amino acids and ions in trypanosomatids is aided by structures called acidocalcisomes (75), which are thought to act as storage compartments for metal ions, inorganic phosphates, basic amino acids, and pH regulation, and might regulate osmotic homeostasis (75), as it reportedly co-localizes with the aquaporin complex (76, 77). Interestingly, the translocation of acidocalcisomes arising from hypo-osmotic induction was mimicked by treatment with dibutyryl cAMP (77). Treatment with the PDE inhibitor dipyridamole also elicited this effect, whereas the AC inhibitor 2-deoxy-3-AMP inhibited translocation of acidocalcisomes (77). The trafficking of the aquaporin-acidocalcisome complex resembles the well-characterized cAMP-PKA-regulated aquaporin translocation in mammalians (78) and could reveal a new, critical role for cAMP and a putative PKA in trypanosomatids. It will be important to reconfirm this observed effect, however, with hydrolysis-resistant analogs of cAMP, (i.e., Sp-8-Br-cAMP or 8-pCPT-cAMPS), because dibutyryl cAMP breaks down to butyrate (which has significant biological activity) and the PDE inhibitors used (76) only slightly inhibit trypanosome PDEs (i.e., IBMX) or have multiple targets (i.e., dipyridamole). Similarly, trypanosome ACs are poorly inhibited by 2-deoxy-3-AMP.
Another area of widespread interest is the possible role of cAMP in flagellar function. Trypanosome flagella are crucial for diverse functions, ranging from cell motility to the trypanosome cell cycle (79, 80). A role for cAMP in trypanosome flagellar function remains speculative, but there are some interesting similarities between trypanosome flagella and the flagella of mammalian sperm (81), in which the participation of cAMP is critical. Sperm motility and male fertility are governed by numerous ACs, PDEs, PKAs, and AKAPs that form a cAMP signaling complex. As described above, trypanosome flagella also appear to contain complete cAMP signaling complexes associated with the PFR structure (81). Could the PFR be a scaffold bringing together diverse players in the cAMP signaling cascade, forming a cAMP microdomain important in flagellar function? And will many of the findings in mammalian sperm be applicable to trypanosomes?
Roles for cAMP: Reinterpreting the Past
We must be cautious with regard to some of the suggested roles for cAMP in trypanosomes. Many of the studies described earlier assumed that inhibitors or activators of mammalian ACs or PDEs would also inhibit their trypanosome counterparts. Although many of these analogs do have an effect on trypanosome proliferation or differentiation, we now know that they might do so in a cAMP-independent manner. Hence, the effects observed may not be due directly to cAMP.
Many of the suggestive roles of cAMP have come from experiments with membrane-permeable cAMP analogs. For example, in the mammalian bloodstream, T. brucei can transform from a rapidly proliferating slender form to a non-proliferating stumpy form. When characterizing an extracellular factor (called Stumpy Inducing Factor or SIF) that caused the transformation from slender to stumpy forms in T. brucei, investigators used a cell-permeable cAMP analog 8-(p-chlorophenylthio)-adenosine-3′,5′-cyclic monophosphate (8-pCPTcAMP) to mimic this transformation in bloodstream form T. brucei (22, 23) mimicking the actions of SIF. This effect was also seen with etazolate, a mammalian PDE inhibitor and a putative trypanosome PDE inhibitor. The transformation was therefore attributed to a cAMP effect. However, more recent work has shown that many membrane permeable analogs {e.g., 8-pCPT-cAMP and [8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate] 8-pCPT-2′-O-Me-cAMP} are hydrolyzed efficiently by trypanosome PDEs (82) (Figure 5⇓). Moreover, the hydrolysis products (the 5′-AMP/adenosine analogs) appeared to be more potent than the cyclic analogs themselves in arresting proliferation and causing the transformation of slender T. brucei into stumpy-like forms (82). Additionally, etazolate promoted the same transformation in vivo with a higher potency than its ability to inhibit PDE activity in vitro, supporting the argument that the effects of etazolate may not be directly owing to effects on cAMP but rather caused by some other effect of this membrane-permeable adenine/adenosine analog.
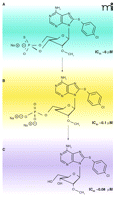
Hydrolysis products of cAMP analogs in trypanosomes have anti-proliferative activity. Membrane-permeable cAMP analogs are hydrolyzed by T. brucei PDEs, and the hydrolysis products (adenosine and 5′-AMP forms) show stronger anti-proliferative activities than the cAMP analogs themselves. These hydrolyzed products cause the conversion of long, slender forms of T. brucei to short, stumpy-like forms. Shown here are the structures and IC50 values for the antiproliferative effects of 8-pCPT-2′-O-Me-cAMP (A) and its hydrolysis products, 8-pCPT-2′-O-Me-5′-AMP and 8-pCPT-2′-O-Me-ado (B and C, respectively). Some of these cell-permeable adenosine analogs could be further developed as potential anti-trypanosomal agents. Data from (82). Used by permission. ©2006, National Academy of Sciences Press.
The nucleotide analog study therefore suggests a need for careful re-interpretation of previous studies (utilizing cAMP analogs) and the conclusions posited regarding cAMP-mediated effects on trypanosome differentiation and function. Some of the numerous effects of these analogs may indeed be cAMP-mediated but others likely arise from non-cyclic metabolites of cAMP analogs. Interpretations are further clouded by the fact that some compounds (like caffeine or IBMX) appear to alter cAMP concentrations in trypanosomes (15, 20) even though none of the trypanosome PDEs are inhibited by these compounds. The same effects should be reinvestigated using hydrolysis resistant membrane permeable analogs, such as the Sp and Rp isomers of cAMP, or analogs such as Sp-5,6- Dichloro-1-β-d-ribofuranosyl-benzimidazole-3′,5′-cyclic monophosphorothioate (Sp-5,6-DCl-cBIMPS). Interestingly, studies with adenosine analogs suggest membrane-permeable adenosine analogs as potent modulators of trypanosome function and, perhaps, raise the possibility of their use as therapeutic agents (82). For example, cell-cycle arrest of the bloodstream form of T. brucei in the stumpy form can lead to lowering of parasitemia in the mammalian host (4, 83).
Conclusions
We are only now beginning to understand cAMP signaling in trypanosomes. Some of the putative roles of cAMP in these parasites require more critical analyses and reinterpretation, but other studies suggest new, relatively unexplored roles for cAMP in these parasites. Our improved molecular understanding of trypanosome PDEs and ACs show substantial differences between them and their mammalian counterparts. Given these differences, it should be possible to develop specific and selective agonists or antagonists for trypanosome cyclases or PDEs. These pharmacological agents could be extremely useful tools to unravel the specific physiological roles of cAMP in both T. brucei and T. cruzi. Some of the roles for cAMP in these parasites appear to be essential for their survival. Hence, these pharmacological agents also hold high promise as potential drugs to treat these devastating diseases that are presently poorly controlled by current drug treatments. Finally, there is also a need to identify effector proteins of cAMP in the various trypanosomatid species. In mammals, our current understanding of cAMP function has expanded as through the further characterization of cAMP effector proteins such as PKA and EPACs; however, cAMP signaling in these parasites may be substantially different from mammals or other eukaryotes. Given the recent advances in proteomic techniques the time is ripe for new searches to identify and characterize cAMP binding and regulated proteins in trypanosomes. This promise of new discoveries with potentially substantial therapeutic benefits should drive research about cAMP signaling in trypanosomes forward in the future.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Joe Beavo received his PhD in 1970 from the Department of Physiology at Vanderbilt University, where his thesis research was carried out with Drs. Joel Hardman and Earl Sutherland. He then moved to the Department of Biological Chemistry at the University of California, Davis, where he worked with Dr. Edwin Krebs. In 1977, he joined the University of Washington as an Assistant Professor of Pharmacology and has been on the faculty there since that time. His research interests have focused on many different aspects of cyclic nucleotide metabolism, particularly as regulated by cyclic nucleotide phosphodiesterases. E-mail beavo{at}u.washington.edu; fax 206-685-3822.

Sunil Laxman received his PhD in fall 2006 from the Department of Pharmacology at the University of Washington, Seattle. His thesis research was carried out with Dr. Beavo, where he studied the mechanisms of cAMP signaling in trypanosomes. He is currently a postdoctoral scholar at the Department of Biochemistry at the UT Southwestern Medical Center at Dallas. He is broadly interested in signaling in relation to metabolism. E-mail sunil.laxman{at}utsouthwestern.edu

