BEYOND AMYLOID: The Next Generation of Alzheimer’s Disease Therapeutics
Abstract
The precise pathological events that cause cognitive deficits in Alzheimer’s disease remain to be determined. The most widely held view is that accumulation of amyloid β peptide initiates the disease process; however, with more than eighteen amyloid-based therapeutic candidates currently in clinical trials, the targeting of amyloid alone may not be sufficient to improve functional deficits over the course of the disease. Alternative targets, such as the tau protein and apolipoprotein E, have thus been increasingly investigated, and in the future, therapeutic strategies will likely address events that are upstream of a more broadly construed pathological cascade that includes but is not limited to the generation and accumulation of amyloid β. Consideration of such events provides the basis for an “indirect amyloid hypothesis,” for which data are beginning to emerge. Although it is clinically defined by simple post-mortem criteria, Alzheimer’s disease likely has a complex etiology, and effective treatments for this disease will become ever more urgent as the world’s population ages.

Introduction
The most widely accepted view of the pathogenic events that culminate in Alzheimer’s disease (AD) centers around the proteolytic generation of the amyloid β peptide (Aβ) and its accumulation in the brain [for review, see (1, 2)]. The Aβ cascade hypothesis holds that the accumulation of aggregated Aβ species is the key initiating event in the development of AD, although the precise species of Aβ that drives neurotoxicity is one of the many uncertainties that remain to be resolved. This hypothesis continues to be controversial (3) and has yet to be formally tested; however, it is supported by compelling genetic evidence. Mutations in either the gene that encodes the amyloid precursor protein (APP) or in genes that encode components involved in the processing of APP [e.g., presenilin-(PSEN)-1] can lead to familial AD. Indeed, such mutations either increase the generation of Aβ42, the more amyloidogenic form of Aβ, or increase the tendency of Aβ to aggregate, and along with triplication of the region of chromosome 21 that encodes APP, they account for all known cases of inherited AD (4). Moreover, variation in the gene that encodes apolipoprotein E (APOE) has been linked to AD, and there is good evidence that APOE genotype influences amyloid deposition either directly or by affecting the rate of Aβ clearance from the brain (5, 6). Finally, several other forms of amyloidosis are associated with progressive neurodegeneration, such as British Dementia, in which case the analogous precursor protein carries a C-terminal extension that aggregates and deposits into amyloid plaques associated with disease progression (7). The obvious parallel between the pathogenesis of British Dementia, which depends on the generation of a mutant amyloidogenic peptide, and AD provides further support for the Aβ cascade hypothesis.
In addition to genetic data, a multitude of studies have shown that Aβ can be toxic to neurons in culture, and several studies have recently shown that specific oligomeric Aβ species, when injected into rodents, cause memory deficits (8–10). Indeed, increasing evidence indicates that overexpression or accumulation of various Aβ species consistently results in synaptic dysfunction in mice expressing mutant APP alone or in combination with presenilin transgenes (11–16). Consequently, many pharmaceutical companies have invested in the discovery of small-molecule and biologic agents that target specific components of the amyloid cascade (17, 18). Over seventy-five compounds are in clinical development for the treatment of AD, of which greater than 80% are based on either amyloid-specific mechanisms or on neurotransmitter pathways (Table 1⇓; Figure 1⇓); the latter are primarily palliative, aimed at affecting learning and memory. Here, we focus on therapeutic discovery that has not been primarily driven by the amyloid hypothesis. We also discuss how modern genome-wide small interfering RNA (siRNA) screens and molecular profiling can be employed to further delineate pathways in AD development, independent of their potential role in Aβ generation.
Non-amyloid–based drugs in clinical development for Alzheimer’s disease.

Classes of compounds in clinical development for the treatment of Alzheimer‘s disease. Numbers in parentheses represent number of drug companies that are pursuing the given drug class. (See http://clinicaltrials.gov/).
Beyond the Amyloid Hypothesis
The central nervous system has proved to be a challenging area for drug discovery, which undoubtedly reflects the complex nature of the brain. Likewise, it should not be a surprise to learn that the majority of clinical development candidates fail not only because of safety or tolerability issues, but also because of flawed hypotheses that may require more than a decade’s worth of investigation prior to testing in patients (19). These considerations, and recent disappointing results from phase III trials of the anti-amyloidogenic agent tramiprosate, underscore the necessity of exploring therapeutic targets for Alzheimer’s disease in a broader context than that offered by the amyloid hypothesis.
Despite the wealth of evidence indicating a central role for Aβ in the etiology of AD, there are significant problems with a simplistic hypothesis in which aggregated Aβ species directly cause neuronal dysfunction and lead to the clinical phenotype. First, there is the well-documented lack of correlation between the extent of amyloid pathology in the brains of AD patients and clinical progression (20). Indeed, a significant number of asymptomatic individuals show extensive, if diffuse, amyloid deposition at autopsy (21). A further complication is that at least in early-stage AD, neuronal loss and amyloid deposition occur in different brain regions (22). Both issues have yet to be satisfactorily explained within a minimal amyloid cascade model, although biochemical studies have indicated that soluble Aβ species may display a more convincing link to disease progression (23). A further problem with a simple interpretation of the amyloid cascade model is that mice expressing mutant APP transgenes either alone or with a PSEN1 mutant transgene develop extensive amyloid disposition but fail to show frank neuronal loss (24). Indeed, even the extent of synaptic loss varies markedly among different APP- and PSEN1-expressing lines (24). As a result, it seems difficult to argue that exposure to disease-relevant levels of Aβ species directly causes acute neurodegeneration in vivo. Consistent with the absence of neurodegeneration, mouse models that overexpress APP (with or without PSEN1) fail to develop intracellular neurofibrillary tangles (NFTs), which next to amyloid plaques is a primary pathological feature of AD.
All of these concerns might be addressed by modification of the amyloid cascade hypothesis, such that accumulation of Aβ species may initiate the disease process, but a secondary pathogenic event is required for subsequent neurodegeneration. Dysfunction affecting the microtubule-associated protein tau (MAPT), which gives rise to NFTs, is an obvious candidate for this secondary event. NFTs correlate well with clinical progression in AD, and transgenic mice that express mutant MAPT protein and develop widespread NFTs almost inevitably have neuronal loss in affected brain regions (20, 24).
The potential consequence of this modified scheme is that strategies that target Aβ in order to prevent or clear amyloid deposits could prevent the onset of disease but would have limited benefit in patients with established clinical symptoms. Indeed, autopsy studies in patients treated with the Elan Pharmaceuticals vaccine that elicited robust antibody titers against Aβ peptide indicate effective clearance of amyloid plaque pathology; in contrast, the tau pathology in these patients showed no evidence of reduction (25, 26). Twelve months after immunization, patients displayed little clinical benefit (27). These results are consistent with our modified hypothesis, according to which symptoms of neurodegeneration would reflect a point or event in AD progression where Aβ reduction alone may have little effect.
A further complication to the simple, linear amyloid cascade hypothesis comes from indications of molecular mechanisms that indirectly influence amyloid production and likely contribute to the neurodegenerative process in AD. One example comes from a recent study in which hypoxia was reported to cause upregulation of the enzyme that cleaves APP and generates the N terminus of Aβ [i.e., the beta-site APP-cleaving enzyme (BACE)] (28). Along similar lines, brain injury resulting from almost any insult will induce Aβ deposition, perhaps by increased expression of acute phase proteins, including APP and APOE (29, 30). As a result, even if the amyloid cascade hypothesis is fundamentally correct, the clinical symptoms in AD may result from a complex interplay of Aβ-induced neurodegenerative processes along with insults that indirectly influence Aβ production and/or deposition. In turn, these variables would complicate treatment with anti-amyloid strategies, particularly in subsets of patients where the Aβ-driven component of the disease is relatively minor.
Palliative Therapeutic Modulation of Neurotransmitters in AD
The palliative treatment of AD has been practiced for over a decade in the form of AChE inhibitors and glutamate receptor modulators (31). These approaches target the symptomatic deficits in the disease and have significant but modest clinical benefit in specific patient groups. Whether these agents effectively modify disease course continues to be debated at the epidemiological level (32), but there is no compelling evidence that they fundamentally change the underlying disease pathology. Nevertheless, second-generation neurotransmitter strategies may provide a means of testing the indirect amyloid approach. For example, muscarinic agonists not only promote cognition directly, but also inhibit the development of amyloid and tau pathology in a model of AD based on triple transgenic (i.e., APP-, PSEN1-, and tau-expressing) mice (33). Although the underlying mechanism is not clear, the results imply that muscarinic compounds may thus influence the etiology of AD as well as provide symptomatic benefit (34). Several companies are investigating activators of nicotinic acetylcholine receptors and AMPA receptors; however, the potential of these therapeutics to modify the underlying disease pathogenesis is less clear.
Tau—the other “Amyloid”
The identification of pathogenic mutations in the tau-encoding gene (i.e., MAPT) in patients with frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) demonstrates that tau dysfunction can be sufficient to cause neurodegeneration (35, 36). Furthermore, a common MAPT haplotype (H1) is a major risk factor for progressive supranuclear palsy and corticobasal degeneration, two sporadic tauopathies that lack highly penetrant pathogenic MAPT mutations (37, 38). These genetic findings, along with the established correlation between NFT pathology and clinical progression, corroborate the important role that tau plays in the development of AD.
The nature of the MAPT mutations found in FTDP-17 patients suggest that tau-associated neurodegeneration arises either from a conformational change in tau, leading to aggregation, or from tau dysfunction, leading ultimately to microtubule instability (35, 36, 39). Hyperphosphorylation is a common feature of the tau species incorporated into neurofibrilliary lesions in all of the tauopathies, including AD, and hyperphosphorylation has also been shown to block tau binding to microtubules, potentially resulting in a loss of microtubule stability (3). As a result, reported tau-based drug discovery has focused on: i) preventing the accumulation and aggregation of pathological tau species (40, 41); ii) preventing microtubule destabilization that may occur as a result of tau dysfunction (42); and most commonly, iii) inhibiting kinases that have been linked to tau hyperphosphorylation (43–46).
Lessons from Risk Factors in Late-Onset AD
The clearest genetic risk factor for late-onset AD (LOAD) is the presence of the ε4 allele of the APOE gene (47). Although individuals homozygous for the ε4 allele have fifteenfold the risk of AD compared to individuals homozygous for the ε3 allele, mechanistically there has been a paucity of data leading directly to traditional druggable targets. However, scientists at the Gladstone Institute have recently identified potential targets for drug intervention based on disruption of protein conformational structure (48). The difficulty in identifying tractable targets based on APOE4 biology is linked to the complexity of its involvement in brain lipid metabolism and by the incomplete penetrance of this genetic risk factor in the disease. A further complexity is that APOE genotype clearly influences Aβ deposition, even if only indirectly. Elderly individuals with the ε4/ε4 and ε4/ε3 genotypes display greater amyloid pathology regardless of whether they are diagnosed with AD, another neurodegenerative disease, or are unaffected at time of death (7). The potential complexity of the role of APOE in AD raises a particularly interesting concern: If APOE offers us the best chance for elucidating genetic risk factors for AD, then how realistic is it to expect to unravel the mechanisms of less penetrant risk factors? The question is daunting, and answers will likely require genome-wide expression data to identify pathways for exploration.
Biomarkers as Clues to AD Pathology
Fluid biomarker studies of cerebrospinal fluid (CSF) from patients with AD have also identified a number of pathways that may provide clues to therapeutic intervention. In one study, using two-dimensional gel electrophoresis of CSF samples from patients (49), a panel of proteins that distinguish AD from control with an accuracy of 94% was identified. These protein markers are predominantly involved in Aβ transport (e.g., APOE), inflammation (e.g., complement proteins), or protease inhibition , and neuronal membrane proteins such as neuronal pentraxin receptor. Other studies have similarly identified members of the complement cascade as well as proteins of the folate-homocysteine cycle (50–52).
Indeed, many prospective studies now confirm that elevated plasma homocysteine substantially increases risk for dementia in the elderly. It is not clear if plasma homocysteine, a common risk factor in many diseases (e.g., atherosclerosis), contributes to AD pathogenesis or simply marks it. Sontag and coworkers (53) have recently shown that disruption of homocysteine metabolism in mice affects both amyloid and tau pathology in rodents. Intriguingly, this disruption decreases methylation of phosphatase 2a, a phenomenon that is also seen in AD tissue. Studies on AD fluid biomarkers, particularly at different stages of the disease and in at-risk populations, will inevitably yield many further clues. The ongoing government–industry sponsored Alzheimer’s Disease Neuroimaging Initiative (ADNI; http://www.loni.ucla.edu/ADNI), inspired by the late Leon Thal, will provide a unique resource to qualify some of these biomarkers and assess their functional relevance to disease pathology and neuropsychiatric endpoints.
Beyond Amyloid as Direct Target
Regardless of whether it is the direct cause of neurodegeneration, Aβ deposition is a relatively specific hallmark of AD, and multiple pathogenic mechanisms most likely contribute to its accumulation. Thus, an attractive new area for investigation is to identify processes that lead indirectly to increased amyloid deposition.
Cholesterol Homeostasis and Lipid Signaling
Cholesterol homeostasis is of interest (54) because of the fundamental role APOE plays as the major cholesterol carrier in the brain. In addition, hypercholesterolemia has been implicated as a risk factor for AD (55), and membrane cholesterol promotes Aβ production, whereas Aβ40 inhibits cholesterol synthesis (56). Epidemiological studies indicate that cholesterol-lowering medications such as simvastatin may be associated with a reduced risk of dementia in the elderly (55).
Given the tremendous therapeutic success of modulating peripheral cholesterol for cardiovascular disease, one can imagine that there are also additional safe points of intervention into brain lipid metabolism. Among targets that have been proposed are acyl-CoA:cholesterol acyltransferase (ACAT), inhibition of which reduces the pool of brain cholesterol esters and lowers Aβ in transgenic mice. The nuclear receptor LXRβ, which binds oxysterols and thereby stimulates the transcription of genes involved in cholesterol efflux from the brain (e.g., APOE and ABCA1), is also promising. Specifically, LXRβ activation by synthetic ligands increases ABCA1 levels and reduces amyloid load in transgenic mice (57, 58). The low-density lipoprotein receptor–related protein (LRP) appears to promote Aβ formation in murine models (59); inhibition of the LRP–APOE interaction might thus positively affect both APOE and amyloid pathways. Additional targets include the cytochrome P450-(CYP)-46 enzyme, exclusively expressed in the brain, which eliminates brain cholesterol by oxidation to 24-hydroxycholesterol, which diffuses out of the brain. In mice, CYP46 is required for cholesterol homeostasis and proper synaptic function (60), and its pharmacological activation could conceivably inhibit amyloid production, although this hypothesis remains to be tested. Future interest could also include the oxysterol binding proteins (OSBPs), an intriguing family of lipid transporters that bind oxysterols and modulate cell signaling and membrane trafficking (61). Although each of these potential targets faces significant hurdles, the field of brain cholesterol homeostasis exemplifies the indirect amyloid approach.
Energizing the Brain in AD
Another emerging field is brain energy homeostasis. AD patients show profound reductions in brain glucose uptake and metabolic activity early in the disease process (62), and mitochondrial dysfunction has long been suspected in AD pathogenesis (63). Conversely, adults with high education, strong linguistic skills, and a mentally active lifestyle seem to be protected from AD (64, 65). Studies in transgenic mice show that levels of Aβ are reduced by voluntary exercise (66) and mental activity (67, 68) , whereas they are raised upon energy deprivation by 2-deoxyglucose (69).
Other data raise the possibility that biochemical pathways regulating brain energy metabolism could be targets in AD. Indeed, rosiglitazone, an agonist of peroxisome proliferator-activated receptor-(PPAR)-γ, has recently shown hints of efficacy in a subpopulation of AD patients (70) by a mechanism that is hypothesized to involve modulation of neuronal mitochondrial function (71).
Other Pathways into AD
There is also abundant evidence linking inflammation, which likely plays a key role in most age-related degenerative diseases, with Aβ production and toxicity (72). Peripheral anti-inflammatory strategies have yielded effective medicines for many conditions, and it thus seems plausible that specific pathways that modulate microglial activation and influence Aβ accumulation or toxicity could be targeted. Finally, a connection between Aβ and cerebral vasculature pathology has long been appreciated, and recent studies identify serum response factor and myocardin as transcription factors, overexpressed in vascular smooth muscle cells in AD patients, that might mediate arterial hypercontractility (73). Preventing the aberrant expression of these and other factors could represent indirect amyloid strategies. These examples are not exhaustive—similar strategies could focus on oxidative stress, elevated autophagy/lysosomal dysfunction, calcium dysregulation, aberrant cell cycle entry, and related pathways.
Genomic Analysis of Risk Factors
Functional genomics offers the opportunity to identify factors leading to amyloid accumulation or toxicity in an unbiased fashion, so that indirect amyloid targets are not overlooked by design (Figure 3⇓). A genome-wide siRNA screen has validated this unbiased approach to factors that regulate APP processing and secretion (74). Among the genes identified in this way was LRRTM3, which encodes leucine-rich repeat transmembrane protein 3, a novel neuronal receptor-like protein that promotes the proteolytic processing of APP by BACE. Although its function is unknown, LRRTM3 is from the same family as the SLIT receptor kinases and the NOGO receptor, critical regulators of axon guidance, synaptogenesis, and neuroregeneration; the NOGO receptor negatively regulates Aβ production in mice (75). Combined with data suggesting a role for APP in synaptic regulation (76–78), along with the emerging concept that AD involves fluctuations in neurological function (79), these genomic data may culminate in a therapeutic strategy aimed at stabilizing synaptic activity. Specifically, one can envision neuronal proteins such as LRRTM3 and the NOGO receptor as targets of early investigation. Likewise, saturation mutagenesis experiments in model organisms will yield new hypotheses drug targets. Paired with appropriately powered human genetics studies and the identification of disease progression biomarkers, research in AD and related areas could experience a profusion of hypotheses, irrespective of the amyloid hypothesis, that will form the bases for future clinical trials.
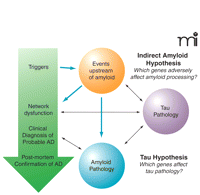
Theindirectamyloid hypothesis of Alzheimer’s disease. Large arrows represent upstream events that may contribute to network dysfunction and subsequently neurological damage. Smaller black arrows are indicative of downstream contributory factors that are likely to be important once the disease is established.
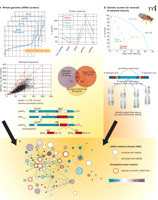
The convergence of two distinct genomic screens that together identify genes that regulate APP processing and amyloid neurotoxicity. A) The development of inhibitors APP proteolysis (i.e., inhibitors of γ-secretase) for the treatment of Alzheimer’s disease has been hampered by mechanism-based toxicities. To identify novel regulators/modulators of APP processing, a whole-genome siRNA screen was conducted in HEK293 cells expressing the NEFV mutant of APP—an engineered mutation that facilitates cleavage and assay of the Aβ42 fragment. Viability and multiple endpoints of APP processing were monitored, including the production of Aβ40, Aβ42, soluble APPβ (sAPPβ), and soluble APPα (sAPPα). Hits were selected based on high-throughput results within two standard deviations from the mean as well as by patterns of inhibition reflecting BACE-like activity (i.e., decrease in Aβ40, Aβ42, and sAPPβ and an increase in sAPPα). [See (74).] B) The expression of human Aβ42 peptides in fruit flies results in decreased lifespan, locomotor deficits, and rough-eye phenotype. These characteristics are not observed in flies expressing the benign human Aβ40 peptide and are exacerbated in flies expressing either a double dose of Aβ42 or aggregation-prone forms of Aβ42. A p-element genetic screen was performed to identify genes that, when overexpressed or disrupted, would rescue flies that express an aggregation-prone form of Aβ42. Several genes were identified that rescue Aβ42-mediated toxicity and are currently under investigation. [See (80).] C) Exploring protein-protein interaction networks reveal a convergence of the data sets indicating that pathways that regulate APP processing may also play a role in mitigating Aβ42 related neurodegeneration.
Conclusions and Future Perspectives
Academia, government, and industry have all made huge investments into research to understand the biology and potential therapeutic points for intervention in AD. The aging population and recent failures of large late-stage clinical studies involving amyloid-directed strategies highlight the sense of urgency. However, emerging evidence about pathways that may lie upstream of amyloid generation sustain efforts to intervene into AD progression. Research focused on mechanisms indirectly related to amyloid production will likely provide the basis for the next generation of targets for AD. However, no validated animal models are yet available to test the efficacy of such approaches to the disease, and proof of concept will involve large and extended clinical studies. This situation highlights the need for further research into disease biomarkers. Such biomarkers cannot realistically be expected to become a surrogate for the disease in the near term, but they will become crucial to establishing markers of central target engagement and will perhaps provide clues to alternate targets for therapeutic intervention. Disease biomarkers are also needed to prioritize research investments into those compounds and approaches that are likely to have the highest probability of efficacy against AD. Thus, an integrated approach to treating AD is essential to success in fighting this disease (Figure 4⇓). This integration will necessitate a comprehensive understanding of the genetics of vulnerable patients, an appropriate use of diagnostics and disease state biomarkers, as well as imaging and neuropsychiatric endpoints to track disease progression and therapeutic outcomes.
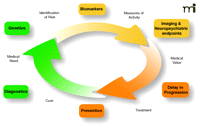
A picture of the future: the Alzheimer’s disease healthcare cycle. This schematic highlights the importance of taking an integrated approach to identification of novel therapeutic targets for Alzheimer’s disease by considering not just the genetic basis of a given target but also its biological context and how biomarkers coupled to clinical endpoints will be needed to measure efficacy to establish medical value to patients and caregivers.
Acknowledgments
We would like to thank Lennart Mucke for critical insights on this review. Also, we would like to thank Luiz Miguel Camargo, Uwe Beffert, and Dianne McDonald-Harmer for their contributions in preparation of this manuscript, as well as Richard Page, David Lomas, and Damian Crowther (University of Cambridge, UK) for their collaborative and pioneering work on the Drosophila amyloid model.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Mark Shearman, PhD, studied biochemistry at the University of Nottingham (UK) and pursued postdoctoral appointments in Japan and Germany. He has played multiple leadership roles since joining Merck in 1992, and in 2006 moved to the new MRL research facility in Boston to establish and lead neuroscience efforts as Executive Director, Neuroscience Drug Discovery. Dr Shearman’s research has advanced understanding of the γ-secretase enzyme complex and its role in Alzheimer’s disease pathophysiology.

Michael Hutton, PhD, completed his doctoral work at University of Cambridge, UK, and subsequently rose through positions in academia. His laboratory, identified mutations in MAPT that cause frontotemporal dementia, demonstrating that tau dysfunction is sufficient to cause neurodegeneration, and went on to elucidate, through the development and use of elegant transgenic mouse models, the roles of tau and the growth factor progranulin in distinct AD-associated processes. Dr Hutton recently moved to Merck Research Laboratories in Boston to head a group that will focus on AD and related neurodegenerative conditions.

Guy R. Seabrook, PhD, studied neurophysiology at the University of Nottingham (UK) and subsequently pursued postdoctoral work in both academia and industry. He joined Merck’s West Point PA facility in 2002 and led the Therapeutic Area Group for basic research into movement disorders. He is currently Head of Department for West Point Alzheimer’s Research and has worked on numerous successful drug discovery projects and development teams. Address correspondence to GRS. E-mail guy_seabrook{at}merck.com; fax 215-652-2075.

