From Bioinformatics to Bioassays: Gleaning Insights into Protein Structure-Function from Disease-Associated nsSNPs
Advances in technology have made large-scale “omic” screenings possible, resulting in an abundance of data that now beg for equally large-scale efforts at interpretation and evaluation of relevancy. One such arena of large-scale analysis involves the scrutiny of single nucleotide polymorphisms (SNPs) (Box 1) found in DNA. Whole-genome sequencing efforts have yielded an enormous number of non-synonymous SNPs (nsSNPs); however, for most of these, the functional consequences of the changed amino acid remain unknown. It is important to distinguish nsSNPs with functional consequences because some cause or contribute to a wide variety of human diseases. As personalized medicine advances, individuals who undergo gene sequencing will want to know whether any nsSNPs found put them at increased risk of disease development. The consideration of nsSNPs only, by definition, excludes other categories of SNPs, such as those in non-coding regions or those that affect the third, or “wobble,” base of a codon but do not encode a change in amino acid. To complicate matters, mounting evidence shows that many non-coding regions are transcribed and affect normal cellular functioning (1), and although mutations in the wobble base may not affect amino acid sequence, they may affect the kinetics of translation (2). Emphasis continues to be placed on nsSNPs, however, as numerous examples linking nsSNPs to disease are already known (3).
Regarding the current strategy of undertaking large-scale screenings, how can disease-causing nsSNPs be more easily (or quickly) identified? The large number of nsSNPs obtained can be prioritized using various computational, predictive methods utilizing parameters that include degree of conservation, biochemical and biophysical properties of amino acids, and protein structure data (2, 3). The ultimate validation is a biological assay that directly measures the consequence of the mutation in question, confirming or discarding the importance of a particular nsSNP to disease.
Knowledge of protein structure-function relationships can aid in the prediction of whether an nsSNP will alter protein function and cause disease. In an interesting twist, a recent comparison of known disease-causing SNPs vs common SNPs (not known to be associated with disease) in eukaryotic protein kinases (ePKs) has revealed new insights into regions of the catalytic core most sensitive to change (4). These findings contribute to our overall understanding of protein kinase structure and function.
The protein kinase catalytic core can be divided into subdomains [enumerated as I–XII in (4)] that are highly conserved across almost all protein kinases (5). These subdomains are involved in binding the phosphate donor ATP (or GTP), binding and orientation of the substrate, or catalytic transfer of the phosphate. The activity of protein kinases is essential to nearly all cellular processes (6); thus, any alteration of kinase activity may have a harmful effect. The protein kinase family is predicted to constitute over 20% of the druggable genome, and protein kinases are already a major category of current therapeutic drug targets (7).
Torkamani et al. (4) catalogued publicly available protein kinase nsSNPs [also see (8)]. Disease-causing nsSNPs were gathered from Online Mendelian Inheritance in Man (OMIM) (9), KinMutBase (10), and the Human Gene Mutation Database (HMGD) (11), and common SNPs, of unknown functional consequences, were gathered from dbSNP (12). The data set included SNPs from a large number of different ePKs; this study derived its robustness from examining the amino acid and structural conservation between these different ePKs. The nsSNPs were aligned with the amino acid sequence of the kinase catalytic core. In some cases, multiple different mutations occurred at the same amino acid residue in a given protein kinase. In order to emphasize the comparison between the different ePKs, a mutated residue was only annotated once within a given protein, regardless of the number of different SNPs affecting the same residue (thus creating a non-redundant set of SNPs). (When the authors speak of a high frequency SNP site, they mean the same conserved site is mutated in multiple different protein kinases.)
Figure 1A⇓ illustrates subdomains I–XII of the catalytic core of protein kinase A (PKA), and Figure 1B⇓ shows the location of amino acids with a high frequency of association with disease-causing nsSNPs [from (4)]; a high frequency was considered to be four or more SNPs (found in different kinases) at the corresponding residue. The ratio of observed SNP occurrences to the number predicted (based on random distribution) revealed a general trend toward higher concentration of disease-causing SNPs within rather than between the subdomains. This finding is consistent with the high degree of conservation used to define the subdomains (5), suggesting that the lesser-conserved intervening regions might be more tolerant of polymorphisms as well.
Another pattern observed was the greater frequency of disease SNPs and the greater number of high-frequency SNPs in the C-lobe (approximately subdomains VI–XII) than in the N-lobe (Figure 1B⇓). The N-lobe is primarily important in binding Mg2+-ATP; the C-lobe binds the peptide substrate and contains the invariant aspartate involved in the phosphotransfer reaction (D166) (5).
Torkamani et al. evaluated each high frequency SNP for known disease associations (variations in type of disease and severity) and considered putative mechanisms for altered kinase activity, based on what is known about the functional role of the involved amino acid (4). Certain general trends were found to hold true across all subdomains. SNPs were seldom in residues directly involved in catalysis, and when SNPs did involve catalytic residues, there were severe phenotypic consequences. Disease-causing SNPs were more often observed in residues that affect substrate binding or perform a regulatory role, often indirectly via allosteric networks. Thus, disease-causing mutations usually alter (i.e., increase or decrease) but do not eliminate kinase activity.
Multiple amino acids are conserved in ePKs that are not found in distantly related prokaryotic kinases, the eukaryotic-like kinases (13). It is noteworthy that Torkamani et al. observed a high frequency of disease-causing SNPs in these regions (4), which are proposed to serve regulatory roles in the ePKs. Subdomains IX–XII harbor several of these ePK-specific residues; in addition to the hydrophobic substrate-binding pocket, several residues from different domains (P207 and E208 of the APE motif, W222, and R280), along with a buried water molecule, were proposed to form an allosteric network that couples the substrate- and ATP-binding regions (13). The high frequency of disease-causing SNPs at E208, W222, and R280 serve to corroborate the importance of these interactions (4).
Some of the broad conclusions from this study are likely to extend to SNPs in other enzymes: 1) few disease-causing nsSNPs are likely to occur in direct catalytic sites; 2) mutant proteins resulting from disease-causing SNPs will retain some altered activity, and 3) affected residues may involve substrate binding and regulation of catalytic activity. Structure-function data on the enzyme in question obviously must be available to apply these predictions.
Going beyond correlative evidence that links a specific nsSNP to a disease state, functional assays that test the effect of a given SNP on protein function—either in vitro, in cells, or in some cases, in animals—can distinguish a harmless SNP from one that is detrimental and help determine its causative contribution to disease. Indeed, such a functional assay has recently been developed and tested for SNPs found in the BRCA2 gene (14). The 3,418 aa BRCA2 protein participates in homologous recombination-mediated repair of double-strand DNA breaks (15). Mutations which result in a truncated BRCA2 protein are associated with the hereditary development of breast and ovarian cancer (16). More than 800 mutations of unknown functional consequence, including nsSNPs, have been reported from sequencing of the BRCA2 gene of patients with a family history of breast cancer [from the Breast Cancer Information Core (BIC) database (17), as reported in (18)]. Many patients are thus left with an ambiguous answer as to their risk for the development of breast cancer even after undergoing genetic screening, so the need to distinguish harmful from neutral mutations in the BRCA2 gene is great.
Kuznetsov et al. developed a functional BRCA2 assay utilizing mouse embryonic stem (ES) cells (14). ES cells completely lacking BRCA2 are not viable (19). The new assay capitalizes on this requirement for functional BRCA2 by attempting to rescue BRCA2-deficient ES cells with mutant BRCA2 sequences. Kuznetsov et al. engineered mouse ES cells in which one BCRA2 allele is disrupted, and the other can be conditionally deleted after the introduction of human BRCA2 (wild-type or mutation-containing) (14). Thus, only cells with an introduced BRCA2 capable of supporting viability survive. These cells, once established, can be evaluated in additional assays to assess BRCA2 functions including enhanced sensitivity to cross-linking and methylating agents and defects in radiation-induced DNA repair and homologous recombination. Using previously characterized mutations, the ability of the assay to distinguish deleterious and neutral variants was validated. The authors suggest their assay can be used to categorize mutations of unknown functionality and that such results may be useful to genetic counselors. Similar assays may be developed to investigate mutations, including SNPs, in other human disease-associated genes that result in a phenotype, provided such a phenotype is detectable in ES cells. An advantage is that such assays can, in some cases, be developed without a complete understanding of protein function.
Single Nucleotide Polymorphisms: Harmless and Deleterious
SNPs: “DNA sequence variations that occur when a single nucleotide (A, T, C, or G) in the genome sequence is altered. Each individual has many single nucleotide polymorphisms that together create a unique DNA pattern for that person. SNPs promise to significantly advance our ability to understand and treat human disease” (20).
nsSNPs: Non-synonymous single nucleotide polymorphisms (nsSNPs) that lead to an amino acid change in the protein product are of particular interest because they account for nearly half of the known genetic variations related to human inherited diseases, as reported in the OMIM and HGMD databases (3). Estimates suggest that there are 67,000 200,000 common (occurrence in >1%) nsSNPs in the human genome (21–23). A “classic” example of a disease-associated nsSNP is that of the T-for-A substitution (Val for Glu) in the beta chain of hemoglobin that results in sickle cell anemia (24–26).
Both of these studies clearly demonstrate that the process of discriminating between harmless SNPs and those associated with disease remains a difficult one. The utilization of computational methods and the development of a functional assay discussed above drew from a large body of information already available. Optimal progress in categorizing uncharacterized SNPs, to the ultimate benefit of the patient, will occur at an interdisciplinary interface involving genomics, bioinformatics, structural biology, and biochemistry.
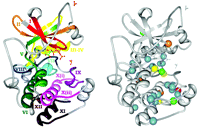
Kinase subdomains and SNP distribution. A. The subdomains PKA (PDB entry 1ATP). Gray residues are intervening loops. Subdomains are numbered by roman numerals and color coded. B. The distribution of kinase disease SNPs. Spheres denote residues with high disease SNP frequencies; red, eight SNPs; yellow, seven SNPs; orange, six SNPs; green, five SNPs; and blue, four SNPs. Reprinted with permission (4).
Acknowledgments
The writing of this article was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
- © American Society for Pharmacology and Experimental Theraputics 2008
References
Joan Cmarik, PhD, is a Staff Scientist in the Retroviral Molecular Pathogenesis Section of the Laboratory of Cancer Prevention at the National Cancer Institute (NCI) in Frederick, Maryland, where her research is focused on the molecular mechanisms of retrovirus-induced leukemia and neurological disease in animal models. Previously at the NCI, Dr. Cmarik carried out postdoctoral research on the regulation of gene expression during neoplastic transformation. She earned her doctoral degree, as a University Fellow, in the Department of Biochemistry and the Center in Molecular Toxicology at Vanderbilt University in the laboratory of Dr. F. Peter Guengerich for her work on carcinogen-induced DNA mutations, during the time when the genetic bases for polymorphisms of human cytochromes P450 were first being discovered. E-mail cmarik{at}ncifcrf.gov; fax 301-846-6164.

