Antifibrillatory Agents and Potassium Channels in the Atria: Pore Block versus Channel Trafficking
Abstract
Atrial fibrillation (AF) is the most common cardiac arrhythmia. The preferred therapy for AF is sustained sinus rhythm control; however, the efficacy of currently used antiarrythmic drugs is limited by adverse side effects resulting from both a lack of ion channel selectivity and nonspecific ventricular activity. The role of the voltage-gated potassium channels in atrial myocyte repolarization and the subsequent control of action potential duration renders them attractive targets for antiarrhythmic drugs in the treatment of AF. Conventional antiarrhythmic drugs generally target the ion permeability of potassium channels. This review discusses the limitations of this traditional approach and introduces, as a novel paradigm for antiarrhythmic pharmacology, the decrease of ion channel cell surface density through the modulation of ion channel trafficking pathways.
Introduction
Atrial fibrillation (AF) is the most common sustained heart rhythm disorder in the United States. It affects over 2.2 million adults and is a major risk factor for increased stroke, heart failure, and cardiovascular morbidity (1). Due to the frequency of AF and its associated morbidity, development of atrial-specific therapies is a major focus of both industrial and academic research efforts (2, 3). The preferred therapy for AF is sustained sinus rhythm control induced by synchronized electrical or pharmacological cardioversion. In addition, catheter ablation has a relatively high success rate, although nearly a third of patients require additional procedures. Some percentage of this additional requirement can be satisfied by dual therapy that combines ablation with pharmacological agents, but the efficacy of existing antiarrhythmic drugs is limited by adverse side effects resulting from a lack of ion channel selectivity and nonspecific ventricular activity (4, 5). Conventional antiarrhythmic drugs generally target the ion permeability of channels. Increasing evidence, however, suggests that functional ion channel density can be modified pharmacologically, in that a drug may both directly block an ion channel and indirectly disrupt normal protein trafficking (6). Emerging studies thus suggest a novel paradigm for antiarrhythmic pharmacology in the control of ion channel cell surface density in atrial myocytes, whereby drug-induced alterations in channel trafficking represent a new mechanism for modifying steady-state ionic current.
Ion Channels in Atrial Fibrillation
Heart rate is tightly regulated and can change in response to metabolic needs and various stressors, such as exercise (7). During normal sinus rhythm, the heart contracts at about 60 beats per minute at rest and up to 200 beats per minute at times of vigorous exertion (7). AF occurs when the control of heart rhythm exits normal sinus node pacemaker activity, resulting in rapid activity in select regions of the atria. This exit from normal rhythm can increase atrial frequency at least seven- to ten-fold, to firing at a rate of 400–600 beats per minute, causing rapid and irregular contracting of the atria (7).
Historically, AF has not been considered a genetic disorder, but rather has been regarded primarily in the context of electrical or structural remodeling caused by re-entrant waves or as a consequence of pre-existing cardiac disorders, such as coronary artery disease, congestive heart failure, and hypertension (7, 8). In the traditional view, electrical remodeling shortens the atrial refractory period, leaving the atria unable to adapt to increases in action potential frequency (9, 10). The atria are, thus, more susceptible to re-entrant waves and rotors—areas of sustained, high frequency reentry. Further, structural remodeling, such as occurs during fibrosis, can lead to the pathogenesis of AF (11). Fibroses can occur in the heart as a result of hypertension, heart failure, or myocardial infarction (11–13). In addition to fibrosis, oxidative stress and inflammation have also been associated with AF (11, 14). However, recent studies have indicated that a genetic component may underlie a subset of AF cases (8). For example, the Framingham Heart Study indicates that offspring of AF parents are at an increased risk of developing AF (8, 15, 16). Regardless of the cause of AF, fibrillatory events generally are initiated in areas of electrical dysfunction in the atria (7).
The normal electrical response in the atria occurs through the coordinated opening and closing of voltage-gated ion channels, culminating in the atrial action potential (17). Depolarization of the plasma membrane results in the influx of sodium, mainly through the cardiac-specific sodium channel Nav1.5, which initiates action potential generation (7, 18). Following sodium influx, calcium entry, mainly through L-type calcium channels, is the trigger for excitation-contraction coupling. The efflux of potassium through two voltage-gated potassium channels, Kv4.3, which underlies transient outward current (Ito), and Kv1.5, which underlies ultrarapid delayed rectifier (IKur) (7, 18), initiates repolarization. Ito is responsible for the early phase of repolarization, where-as IKur is important during early and plateau phase repolarization. Delayed repolarization, following the plateau phase, occurs via the coordinated opening of KCNQ1/KCNE1, underlying the slow out-ward current (IKs), and KCNH2 or hERG2, underlying the delayed rectifier current (IKr) (7, 18). Finally, opening of KCNJ2, underlying the inward rectifier current (IK1), and opening of Kir3.1/3.4, generating the muscarinic potassium current (IKACh), hyperpolarize the plasma membrane and contribute to the shaping of the atrial action potential (7, 18). The ensemble average of all these channels, responding to membrane potential changes in concert, shapes the complex atrial action potential.
Mutations in atrial ion channels or ion channel-associated proteins that alter functional activity can change the shape or duration of the action potential and subsequently the effective refractory period, thus making patients more susceptible to AF. The first reported case of a genetic mutation in an ion channel causing atrial fibrillation was in 2003, when Chen et al. described a missense mutation in the KCNQ1 gene, which encodes the channel that underlies the repolarizing IKs current (19, 20). Although some mutations in KCNQ1 result in loss of function and ventricular long QT syndrome, the S140G KCNQ1 mutation described by Chen et al. is predicted to cause a gain-of-function effect, shortening the atrial action potential duration and increasing the probability of AF occurrences via a re-entrant loop mechanism (19, 20). Re-entrant AF also occurs via gain-of-function mutations in other potassium channel subunits, including KCNH2, KCNJ2, and KCNE2 (8, 21–24). The KCNH2 (hERG) and KCNJ2 (Kir2.1) mutations occur in the ion-conducting alpha subunit and affect IKr and IK1 directly, whereas mutations in KNCE2, encoding a potassium channel β-subunit, alter the ion-conducting properties of I Kr, IKs, and If (8, 21–24). Although these mutations shorten the atrial action potential duration (APD), thereby promoting reentry and AF (7, 8), they can also affect ventricular APD, resulting in ventricular tachycardia and sudden death.
Loss-of-function mutations in other ion channels or related proteins have been implicated in AF (8, 25, 26). Recently, a null mutation in Kv1.5, underlying IKur, was found in a single individual with idiopathic AF (26). This mutation was dominant negative, failing to evoke IKur current and undermining wild-type channel activity (26). Functionally, this effect prolonged APD and led to early afterdepolarizations, increasing the risk for developing stress-evoked AF. Similarly, a point mutation in KCNE1 (S38G), encoding an auxiliary potassium channel β-subunit, has been linked to AF susceptibility, most likely due to the induction of early afterdepolarizations or re-entrant loops (7, 25). Loss-of-function mutations in the sodium channel α-subunit gene, SCN5A, prolong action potential duration and present predominantly as AF, although ventricular tachycardia with Brugada syndrome has also been described (27–29). Whereas these mutations do not represent all loci that may evoke aberrant function and AF, the accumulating data clearly implicate ion channels as therapeutic targets and emphasize the potential value of modulating ion channels in the treatment of AF.
Strategies for the Treatment of AF
Antiarrhythmic drugs that modulate the activity of ion channels are not new but have fallen out of favor due to potential pro-arrhythmic side effects. At this time, one of the front-line treatments to reduce AF symptoms is catheter ablation (30, 31). In this treatment, a catheter is threaded onto the heart and the arrhyth-mogenic region is destroyed. Using this technique, approximately 50% of patients remained asymptomatic without the use of additional pharmacologic agents, whereas an additional 25% required pharmacologic treatment following ablation to maintain normal sinus rhythm (31–33). Despite this success rate, nearly 30% of patients required a second procedure and multiple patients suffered from major complications (31–33). There is no evidence that ablation has any effect on the patient’s risk of stroke or mortality (31–33). Thus, the long-term effectiveness of this treatment remains to be fully determined.
Because AF is a disease of excitability and occurs via abnormalities in the ionic currents underlying the atrial action potential, pharmacologic approaches targeting either atrial-specific ion channels or a combination of ion channels have also been explored (6, 18, 34, 35). Indeed, treatment with ion channel blockers to maintain normal sinus rhythm is the preferred clinical method for the treatment of AF; however, this method is not without its problems. At present, there is no drug in clinical use that does not exhibit significant negative side effects. For example, amiodarone, an effective drug for the treatment of AF, exhibits such side effects as hepatotoxicity, peripheral neuropathy, pulmonary toxicity, as well as many others (35, 36). Quinidine and flecainide, class I antiarrhythmics rarely used in the clinic today, increase mortality via torsade de pointes (35, 37). Finally, the class III antiarrhymic drugs, which block atrial potassium channels, exhibit very little atrial specificity, and similar to class I antiarrhythmics, can be pro-arrhythmic at higher doses (35, 38). Given these adverse side effects, it remains necessary to develop novel, ventricular-sparing blockers of atrial ion channels.
The role of the voltage-gated potassium channels in the repolarization of the plasma membrane renders them attractive targets for antiarrhythmic drugs in the treatment of AF. Perhaps one of the most attractive targets of the voltage-gated potassium channel family is Kv1.5, which underlies the IKur current in the atrial action potential. Despite detection of Kv1.5 protein in the ventricle (39), IKur current has been described in the human atrium only (18, 39–41). Further, it is the main repolarizing current in the atrial action potential and is altered in certain pathophysiologic states (10, 40). For example, Kv1.5 channel protein levels are downregulated in cases of persistant or paroxysmal AF (42), and chronic pulmonary hypertension (43, 44). Because of its atrial-specific expression and its role in disease, Kv1.5 is thus a promising target for the development of new antiarrhythmic drugs.
Over the past fifteen years, Kv1.5 has been the subject of several hundred publications as well as over fifty patents by researchers from both academia and industry (35). Traditional antiarrhythmic drugs, which included the class I antiarrhythmics such as quinidine and propafenone, as well as the class III antiarrhythmics such as ambasilide, amiodarone, and dofetalide (35), have been shown to inhibit Kv1.5. However, due to their lack of specificity and extensive side effects, including ventricular tachycardia and torsade de pointes, these compounds have not proven useful for the treatment of AF. Thus, there has been a push to further understand mechanisms of Kv1.5 block and develop small-molecule inhibitors that minimize extra-atrial actions. Recent work in both sectors has focused on developing Kv1.5-specific blockers for the treatment of AF (2, 3). These drugs have been designed to block Kv1.5 via one of three distinct mechanisms: pore block; allosteric modulation; and most commonly, use- dependent/open channel block (35).
Perhaps the most promising compounds in clinical development for the treatment of AF are those that are not necessarily specific Kv1.5 blockers but nevertheless show atrial selectivity and efficacy (35). Two of these compounds, AVE0118 and AVE1231, are bisaryls developed by Aventis Pharma/Sanofi-Aventis (35). Specifically, they have been shown to inhibit Kv1.5, Kv1.3, Kv2.1, and Kv4.3, with less effectiveness at blocking hERG (17, 35, 45–47). This block of the atrial channels has been shown to prolong action potential duration and reduce vulnerability to reentrant AF occurrences (48–50). Additionally, Merck has had success with DPO-1, a selective Kv1.5 blocker that increases atrial action potential duration, with very little effect on ventricular action potential duration (35). DPO-1 is limited, however, by poor oral bioavailability (35). Icagen has developed several potent blockers of Kv1.5 in vitro, but poor bioavailability has hampered their progress into the clinic (51). Another drug, vernakalant hydrochloride (RSD1235), developed by Cardiome/Astellas Pharma, is a use-dependent blocker of Kv1.5 (52–55). However, at resting potentials, RSD1235 has been shown to also be a potent blocker of Nav1.5 (55), thus limiting its clinical efficacy. This list of compounds, although not extensive, highlights the fact that novel blockers of Kv1.5, compounds with both atrial selectivity and clinical efficacy remain elusive, re-emphasizing our need for new potential therapeutic strategies or targets.
Ion Channel Trafficking as a Target in AF
Historically, antiarrhythmic therapies have been designed to alter conduction through voltage-gated ion channels. More recently, however, a number of such drugs have been shown, upon chronic administration, to function by decreasing functional ion channel density as well as by affecting channel conductance per se (56–60). This dual effect has been best characterized for hERG, where commonly prescribed antiarrhythmic drugs, such as ketoconazole and fluoxetine, reduce hERG density by at least 50% after 48 hours of treatment (56, 57, 61, 62). However, other drugs, such as pentamidine and probucol, reduce hERG cell-surface expression with no apparent effect on ion conductance (56, 63–65). Although these studies show that the role of drugs modulating channel surface density must be interpreted with care and with regard for safety, they do not mitigate the important finding that channel trafficking and pore function represent pharmacologically distinguishable routes for considering AF treatment. Physiologically, reduction in cell-surface potassium channel levels, resulting in decreased current densities, can prolong the cardiac action potential and induce potentially lethal long QT phenotypes (56–58, 66).
Research into the therapeutic value for antiarrhythmic agents that affect channel trafficking has focused almost exclusively on hERG channel blockers that stabilize misfolded channels and rescue hERG trafficking mutants. In addition to drug-induced arrhythmias, over 200 naturally occurring mutations have been identified and shown to cause long QT syndrome due to retention of hERG in the ER (long QT syndrome type 2) (60, 67). Interestingly, treatment with certain antiarrhythmic drugs, such as E4031, rescues the trafficking deficit for nearly 70% of these mutant channels by stabilizing the misfolded protein (60, 67, 89). Despite these clear effects of increasing hERG channel surface density, it is likely that rescue occurs by facilitation of quality control mechanisms that monitor and assist in the folding process to avoid the production of non-native structures and is not directly related to the mechanisms of anterograde trafficking. Recently, a few studies have investigated the cellular machinery controlling hERG stability and trafficking. Apparently, hERG forms a large macro-molecular chaperone complex, including Hsc70, Hsp90, calnexin, and FKBP38, to facilitate folding, maturation, and hERG export from the ER (59, 67–69). Trafficking of hERG from the ER to the plasma membrane is dependent upon interactions with two small molecular weight GTPases, Sar1 and Rab11b (70). These proteins and other elements of the hERG trafficking machinery may thus be relevant to the development of novel therapeutics for the treatment of cardiac arrhythmias.
It is clear that investigations into the precise mechanisms of ion channel transport and also into general protein trafficking mechanisms in the heart have lagged behind comparable processes in other tissues, such as neuronal systems. Interest in the mechanisms of trafficking in non-polarized cells has been sparked, in part, by newly recognized subcellular compartmentalization on the nanometer scale. For instance, it has been shown that multiple potassium channel isoforms localize to lipid-rich microdomains, which may act to stabilize channel proteins in the plasma membrane (71–78). However, there are large gaps in our knowledge of the targeting and trafficking mechanisms for ion channels in the heart.
As mentioned above, Kv1.5 (KCNA5) has emerged as a promising pharmacologic target for treatment of AF. New studies elucidating some of the pathways and proteins involved in Kv1.5 trafficking reveal potential targets for the therapeutic control of channel surface density. The dominant-negative mutation in Kv1.5 that has recently been reported in a familial case of AF (26) arises from a premature stop codon after the S3 transmembrane segment. Interestingly, aminoglycoside-induced translational read-through of the allelic stop codon partially restores channel function and increases current density. Similar to the discussion of the hERG channel above, this translational read-through highlights the potential for pharmacological rescue of disease-causing channel mutations and indicates that steady-state expression levels of Kv1.5 in cardiovascular tissues modulate cellular excitability. The importance of ion channel density is further evidenced by reports showing that Kv1.5 overexpression in rat myocytes results in a phenotype similar to short QT syndrome (79). Conversely, in cases of persistent and paroxysmal AF, there is a decrease in Kv1.5 protein levels as well as a reduction in current density (42, 80). It remains unclear if the reduction in Kv1.5 current density is causative or an effect of AF events.
Work to understand the machinery involved in potassium channel trafficking in the heart has been sparked by a greater appreciation of the dynamics of channel movement and stability. Initial studies indicated that Kv1.5 internalizes in a dynein- and microtubule-dependent manner (81). Recently, we described for the first time the dynamic trafficking of Kv1.5 in atrial myocytes (82, 83). Specifically, a population of Kv1.5 originating on the cell surface internalized to early endosomal compartments, followed by recycling back to the plasma membrane in a Rab4- and Rab11-dependent manner (Figure 1⇓) (82). Disruption of this machinery, using dominant-negative constructs for both Rab4 and Rab11, decreased steady-state surface levels of Kv1.5. These results were confirmed in a separate model, where both Rab4 and Rab11 were shown to be involved in Kv1.5 trafficking (83). Thus, in cases such as persistent and paroxysmal AF where there is a decrease in current density, the use of agents designed to shunt Kv1.5 away from a degradative pathway and into a recycling pathway could prove effective in restoring current density and preventing AF events.
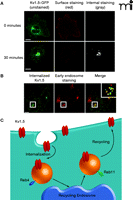
Trafficking of the Kv1.5 channel in HL-1 atrial myocytes. The Kv1.5 channel was constructed as a green fluorescence protein–fusion product in which a single GFP is inserted into an extracellular domain of Kv1.5; the GFP sequence functions as a single epitope in the immunostaining of expressed Kv1.5–GFP described here. (All scale bars are 5 μm.) A. Internalization of Kv1.5–GFP can be monitored as redistribution of green fluorescence over time [left panel; cells following 0 (top) and 30 minutes (bottom) incubation at 37oC are respectively shown]. Because the cells shown were incubated in the presence of a non-fluorescing primary anti-GFP antibody that does not affect trafficking, co-incubation of the cells at 0 or 30 minutes with a saturating red-fluorescing secondary antibody (middle panel) indicates the population of Kv1.5–GFP that remains accessible at the cell surface at the given time; subsequent fixation, permeabilization, and incubation of the cells with a gray-appearing antibody (i.e., non-red; right panel) identifies Kv1.5–GFP that has been internalized from the cell surface. B. Confocal imagery establishes that internalized Kv1.5 can be trafficked to early endosomes. The internalization assay was performed for 30 minutes as described above. Cells were fixed, permeabilized, and stained for internalized Kv1.5 as well as for the early endosomal marker EEA1. The merged image shows co-localized of channel with EEA1 as indicated by yellow punctae. C. The schematic represents the Kv1.5 internalization and recycling pathway. As verified by the confocal imagery with internalized Kv1.5 and shown in the schematic, trafficking of Kv1.5 is both Rab4- and Rab11-dependent. (See text for details.)
As is seen in the case of drugs that target hERG, it is possible that prototypical Kv1.5 blockers may have multiple actions, affecting channel trafficking in addition to altering the biophysical properties of the channel. We have recently discovered that the antiarrhythmic drug quinidine triggers the internalization of Kv1.5 in a dose-, time-, temperature-, and calcium- dependent manner (unpublished data, S Schumacher and J Martens). This internalization was reversible, so that Kv1.5 current density was restored back to normal levels upon washout of quinidine. Interestingly, the quinidine-induced internalization was isoform-specific. For example, Kv4.2 and Kv2.1, which are also blocked by quinidine, did not undergo internalization when treated with maximal doses of drug. Further, Kv1.5 current was blocked but channel internalization was not enhanced in the presence of quinine, the diastereomer of quinidine, demonstrating the stereospecificity of this effect. Moreover, these observations provide additional evidence that ion pore block is separable from drug-induced trafficking effects (Figure 2⇓), suggesting that rational drug design might allow for the manipulation of trafficking pathways without affecting channel function. Equally important, these new data suggest that specific agents can acutely modulate ion channel density, which extends their therapeutic potential but at the same time gives further credence to concerns regarding the comprehensiveness of current ion channel drug safety tests.
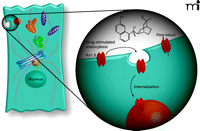
Two distinct mechanisms for acute inhibition of ion current. The classic approach to pharmacological manipulation of channel current is to block or inhibit the conduction of ions through the channel pore (right). As discussed in the text, a second approach is becoming increasingly clear by manipulating the trafficking of channel constituents in order to alter cell surface protein levels (left). One potential therapeutic mechanism would be the antiarrhythmic drug–induced internalization of Kv1.5 to early endosomal compartments. Certain drugs may operate through a combination of pore block and triggered internalization/redistribution of channel proteins.
Future Challenges
Future challenges will arise in the development of novel antiarrhythmic therapies. For example, the question remains as to whether targeting a single channel, such as Kv1.5 for the treatment of AF, would be preferable to a general ion channel inhibitor or traffic-modulating agent. New tools and methodologies are emerging to permit the study of ion channel trafficking and to investigate commonalities among channel isoforms or channel-trafficking pathways. It has been suggested that potassium channels internalize along a common dynein- and microtubule-dependent pathway to endosomal compartments (84), and Kv1.5, KCNQ1, and KCNE1 may all recycle back to the plasma membrane in a Rab11-dependent manner (82, 85, 86). Interestingly, however, they reported a role for serum and glucocorticoid inducible kinase 1 (SGK1) in the membrane recycling of KCNQ1 (81, 83, 85, 86). In contrast, SGK1 has recently been suggested to upregulate Kv1.5 by inhibition of channel ubiquination by Nedd4-2, thus blocking the subsequent degradation in Xenopus oocytes (87). This effect was independent of direct Kv1.5 channel phosphorylation by SGK1. Thus, while similar components are involved in channel trafficking, it appears that unique mechanisms specific to a certain channel or family of channel isoforms may also exist. To further support this possibility, it has been shown recently that Kv4/ KCHIP1 complex trafficking to the cell surface is dependent on interactions with the VAMP7/Vti1a SNARE complex (88). siRNA knock down of either VAMP7 or Vti1a inhibited Kv4/KCHIP1 trafficking to the plasma membrane with no effect on Kv4.2/KCHIP2 or VSVG (88). Together, these data further indicate that ion channel trafficking pathways may be isoform-specific and may be separable from the conventional trafficking of bulk cellular proteins. In any case, identifying similarities and differences in ion channel trafficking pathways and the molecular machinery involved is an important and necessary step for the possible development of new agents that selectively modulate ion conduction and/or the stability of channel protein in the membrane.
Conclusion
AF is the most common cardiac arrhythmia in the United States and is growing exponentially. The invasive cardiac ablation procedure is not universally effective, and existing therapeutic agents designed to target atrial specific channels, such as Kv1.5, appear to lack either atrial selectivity or clinical efficacy. There is, therefore, an unmet medical need for the development of longer-term, ventricular-sparing antiarrhythmic agents. Manipulation of channel-trafficking pathways offers an attractive new paradigm that may provide new novel therapeutic targets for AF. Although much work needs to be done in identifying the precise cellular trafficking machinery and the mechanisms regulating ion channel surface density, recent studies highlighted here suggest that modulation of ion channel trafficking pathways may be an alternative or complementary strategy for treating AF and other potential cardiac arrhythmias.

- © American Society for Pharmacology and Experimental Theraputics 2009
References

Dyke McEwen, PhD, is currently an Application Scientist at Essen Instruments, Inc. in Ann Arbor, MI. His research interests pertain to developing novel methods for long-term live-cell imaging of cells in culture, as well as alterations of ion channel function in disease.

Jeffrey R. Martens, PhD, is Assistant Professor of Pharmacology at the University of Michigan. His interests pertain to the organization, assembly, and dynamics of membrane-delimited signal transduction pathways. Work in his laboratory is focused on understanding the details of Kv channel trafficking, regulation, and pharmacological modulation and how this is all integrated into the broader context of normal cardiomyocyte signaling and the pathogenesis of disease. Address correspondence to JRM. E-mail martensj{at}umich.edu; fax 734-763-4450.

