Antiangiogenic Drugs: Insights into Drug Development from Endostatin, Avastin and Thalidomide
The implementation of an entirely new therapeutic approach can often be highly desired, but it is seldom achieved. One intriguing example of success comes from research into the regulation of angiogenesis. Not only have the scientific advances in this area been breathtaking for their wealth of basic insights into biology, but the data have also been translated into important treatments for cancer and blindness (macular degeneration). These developments also offer potential for further drug and diagnostic opportunities in additional areas of disease. The ongoing journey in this field has also been accompanied by practical lessons for researchers and other professionals engaged in drug development. In this perspective, we describe a number of key aspects learned from the discovery and development of anti-angiogenic drugs, with a focus on endostatin, Avastin, and thalidomide.
Cornerstone Research into Angiogenesis
In 1961, Judah Folkman—a 28-year-old physician, emerging surgeon, and drafted naval lieutenant—was seeking blood substitutes that could be stored for extended periods. Folkman devised a system in which blood substitutes could be assessed for their ability to keep rabbit thyroid glands viable in vitro. One measure of this viability was whether the isolated glands and potential substitutes would support the development of cancer cells into tumors. Folkman and colleagues noted that the tumors that developed in his system stopped growing once they reached a certain limited size. When collected from the in vitro system and implanted into mice, however, the tumors resumed growth and reached massive proportions. Folkman hypothesized that the resumption of tumor growth in vivo was related to the rich vascular bed and blood supply that had been lacking in the in vitro system.
Standard cancer therapy at the time of Folkman’s early work was primarily based on cytotoxic drugs, radiation, and surgery. The cytotoxic drugs then available included mitotic poisons (e.g., Vinca alkaloids), DNA-reactive drugs (e.g., cyclophosphamide), and inhibitors of DNA replication (e.g., 5-fluorouracil), each fraught with problems of efficacy, selectivity, and safety. These inadequacies would later lead the Nixon administration, in the 1970s, to declare “war on cancer” and accordingly influence biomedical research. But in 1967, when Folkman, at the age of 34, became Surgeon in Chief at Children’s Hospital in Boston, he was already motivated by his own lab findings and clinical observations. He was also intrigued by a few generally ignored papers that had earlier hypothesized that angiogenesis represented a promising target for developing better drugs against cancer. His long-time colleague Robert D’Amato would later note that Folkman “...put his patients first and used the laboratory as a weapon for them.”
Folkman observed that the tumors he removed from his patients were “hot and bloody” and highly vascular. It was the relationship between tumors, blood vessels, and blood supply that he chose to attack. In 1971, he published a landmark paper (1), noting that the tumors were typified by new blood vessels. He reasoned that the tumors “...recruited the vessels by sending out some factor that...was diffusible; these diffusible proteins would bring in the vessels, and if you could turn this process off the tumors should stay...small” (emphasis added). This hypothesis is considered today by most students of the field as a “cornerstone” concept (Figure 1), but at the time of its publication, it struck many as heretical.
The search to identify pro- or anti-angiogenic factors as well as antiangiogenic drugs would be long, arduous, contentious, competitive, and expensive. In 1986, Folkman colleague Mike Klagsbrun reported the isolation and purification of the angiogenic factor known as basic fibroblast growth factor (bFGF) (2). In a landmark twenty-three million dollar grant and license agreement with Harvard, bFGF was licensed to Monsanto. The deal included both support and access to Folkman’s discoveries, but three decades of research failed to translate bFGF into an angiogenic therapeutic. Today, at least twenty putative angiogenic factors can be listed (Table 1) (3). Noteworthy among these is vascular endothelial growth factor (VEGF). Originally called vascular permeability factor (VPF) when it was discovered by Harold Dvorak and colleagues at Harvard (4), it was subsequently cloned and expressed by Napoleon Ferrara at Genentech (5). The identification of VEGF and its family of related proteins provided tools to address the key biological hypothesis that had been articulated by Folkman.
The Angiogenic Cascade: Target Identification
Although rapid cell division occurs when tumors are small (1 mm or less), a sustained level of fast growth necessitates an adequate blood supply; tumor cell proliferation is thus balanced against hypoxic cell death. A “little shop of horrors” scenario is established, with the tumor communicating a “feed me, I’m hungry” message that turns on endothelial cell proliferation. This message may in part be carried by bFGF, released by fibroblasts recruited to the inflammatory nidus of the proliferating tumor.
The genes that encode bFGF and VEGF are both activated by hypoxia-inducible factor (HIF), a transcription factor that becomes activated under hypoxic conditions, such as rapidly proliferating cancer cells, and promotes the transcription of a number of genes that support angiogenesis. In the milieu of cytokines and growth factors that begin the process, the expression of angiogenic factors plays prominent roles in stimulating tyrosine kinases and in recruiting differentiating endothelial cells into the leaky neovasculature that feeds tumor growth. At the same time, matrix metalloproteinases carve out paths within the extracellular matrix so that the new blood vessels can be laid down. From the biological hypothesis set forth from Folkman, a variety of players in the angiogenesis cascade have been identified as potential drug targets (3).
In 1990, about the time that interferon alpha provided the first proof of concept that compounds with antiangiogenic activity could treat tumors in patients (6), Judah Folkman and Donald Ingber discovered their first antiangiogenic drug candidate in a sequence of events that was somewhat reminiscent of Fleming’s discovery of penicillin. As Ingber was growing endothelial cells, one of his cultures became contaminated with a fungus that he reasoned might be releasing an antiangiogenic substance (7). Ignoring a general rule that contaminated cultures were not evaluated, Ingber was intrigued and moved ahead with characterization of the antiangiogenic activity, re-discovering fumagillin, a fungal derivative originally discovered in the 1950s. At the time of Ingber’s characterization of fumagillin, Takeda Chemical Industries had been interested in collaborating with Folkman’s lab, and it was opportune that Monsanto’s agreement with Harvard and Folkman was expiring; Takeda had experience and interest in natural products and fermentation technologies. The patent rights to Ingber’s findings were licensed to Takeda. However, in their follow-on studies, it was observed that the prolonged administration of fumagillin resulted in severe weight loss. Subsequently, in 1992, Takeda isolated an active component of fumagillin and synthesized an analog, TNP470, for further clinical evaluation. Unfortunately, after over a decade of research and development, TNP470 was not commercialized for human use, in part due to neurotoxicity, although fumagillin and its analogs continue to attract high interest, particularly for anti-parasitic uses.
Avastin
A different approach to the development of antiangiogenic drugs, as we noted above, began in 1989, in the laboratories of Napoleon Ferrera and colleagues at Genentech (for additional information, see www.gene.com). Having cloned and characterized VEGF (5), important for blood-vessel development and regulation of vascular permeability, the researchers sought to neutralize its angiogenic activity through the use of antibodies. This approach provided not only a means of validating VEGF as a target, but was also advanced in order to develop an antibody-based therapeutic (Figure 2). By 1993, Ferrara and colleagues were able to block VEGF function with a mouse antibody (8, 9), and recombinant technologies were then used to develop the so-called humanized antibody (bevacizumab) that would become marketed as Avastin (10). The success of the project drew from Genentech’s considerable resources and experience with the biology, chemistry, and production of antibodies as drugs.
Genentech also devoted a great amount of resources and time to painstaking clinical analyses. Very early studies with bevacizumab demonstrated significant side effects and mortality in treating brain tumors and lung cancer, but Genentech was able to conduct over sixty phase 2 studies in various cancers, including metastatic colon cancer. The pivotal trial was a large, placebo-controlled, randomized study that demonstrated a five-month prolongation in the median survival of patients treated with Avastin plus irinotecan, 5-fluorouracil, and leucovorin (i.e., IFL) as compared to patients treated with the IFL chemotherapy regimen alone. In addition, progression-free survival (PFS) was increased by more than four months. These and other results, observed when Avastin was added to first-line chemotherapy, were modest in an absolute sense but were the most significant advances ever reported in a randomized, phase 3 study of patients with metastatic colorectal cancer. In 2004, Avastin became the first antiangiogenic drug approved by the US Food and Drug Administration (FDA) (10), initially intended for combination use with standard chemotherapy for metastatic colon cancer; approval for use in non-small-cell lung cancer and breast cancer, and most recently, renal cancer, followed. The cost for Avastin’s research and development was $2.25 billion. From Folkman’s 1972 hypothesis of angiogenesis as key to cancer, it thus took over thirty years to bring the drug to market, and Avastin attests to the fact that making an innovative drug is an expensive and lengthy process. We now turn to an example where time, money, and know-how were not sufficient to ensure product success.
Angiostatin and Endostatin
It naturally stood to reason that if there were endogenous angiogenesis stimulators such as bFGF and VEGF, there must also be endogenous angiogenesis inhibitors. Brem and Folkman (11) described the first endogenous antiangiogenic substance, derived from cartilage, in 1975, and the interplay of pro- and antiangiogenic forces was thus envisioned to determine the balance between controlled growth and malignancy (Figure 3). We now know a number of endogenous compounds with antiangiogenic properties (Table 2).
In 1993, EntreMed, through its sponsored research agreement with Folkman at Children’s Hospital, supported the discovery of two cornerstone endogenous antiangiogenic molecules. The first of these came to light when Michael O’Reilly, working in Folkman’s laboratory, isolated a protein from the urine of cancerous mice that inhibited endothelial cell proliferation and the growth of malignancies (12). This biological derivative of plasminogen, named “angiostatin,” incited great interest, and EntreMed scientists established a pilot scale method for producing a recombinant form of the protein in yeast. The promise of angiostatin caught the imagination of Leon Rosenburg, then President of Bristol Myers Squibb, and by 1996, a strategic deal was struck with EntreMed to bring the drug to market. The industrial partnership was quite noteworthy, with EntreMed receiving over $76 million in potential funding and milestone payments, with significant royalties upon sales. Unfortunately, the great promise of angiostatin was never fulfilled, in part due to its being eclipsed, in 1997, by endostatin, the second of EntreMed’s cornerstone proteins, also discovered by Michael O’Reilly (13).
Endostatin was shown to act on tropomyosin to inhibit endothelial cell growth at the earliest stages of angiogenesis (14), presumably before malignancy and drug resistance could be established (15, 16). Genentech’s Avastin began clinical trials at the same time endostatin was discovered in 1997, and in head-to-head comparisons endostatin was shown to be at least as effective in arresting tumor growth (16). The significance of these comparisons and the promise of endostatin are not often realized today, owing to a variety of events that ensued.
In May, 1998, the New York Times interviewed, among others, Nobel Prize winner James D. Watson (17). In the course of his interview, Watson expressed excitement about Folkman’s pioneering research and the promise of endostatin. The creation of a “buzz” about potentially exciting drugs or technology breakthroughs can result in positive or unrealistic expectations and negative consequences. In this case, the Times drew on comments attributed to Watson, regarding the potential of new drugs “to cure cancer within two years,” for its front-page headline. Media attention and Wall Street enthusiasm for the discovery-stage drug candidate and its developer, EntreMed, became great, but were ultimately premature.
The following November, The Wall Street Journal reported that Folkman’s work was non-reproducible (18), even though researchers, at both the National Cancer Institute (19) and Harvard (20), had in fact verified the findings. As a result of the Wall Street publicity, investors and strategic partners became ultra cautious, and some withdrew support from endostatin, despite its potential, in favor of other antiangiogenic candidates. EntreMed’s financing opportunities disappeared. In early 2003, despite some evidence of early clinical successes, including many patients with stable disease, the high cost of endostatin production and the general “nuclear winter” in the financing of the biotech industry prompted EntreMed to abandon the development of endostatin.
Work on the biological role, mechanisms of action, and analogs of endostatin, however, continued to be pursued by others. Luo Yongzhang and partners at the Chinese company Yantai Medgenn developed Endostar, a soluble and stable endostatin analog containing a nine-residue His-tag and expressed in E. coli (21). Early investigations for manufacturing endostatin had favored a eukaryotic (yeast) expression system, which had appeared to satisfy demands of protein folding and post-translational processing (16). In September 2005, Endostar was approved in China for the treatment of patients with non-small-cell lung cancer. Although Endostar is widely sold in China, its clinical efficacy in the absence of co-administration of other traditional chemotherapeutics has yet to be shown. As exemplified by the progress of Endostar, relative to EntreMed’s history with endostatin, early assessment of methods and costs of manufacturing is essential to drug development.
Thalidomide
The study of angiogenesis in cancer and the search for novel therapies unexpectedly resulted in the resurrection and widespread use of one of the most infamous drugs in the history of therapeutics. In 1992, at the Massachusetts Eye and Ear Hospital, Robert D’Amato was completing his residency. He had become very interested in angiogenesis in diabetic retinopathy and macular degeneration. In a very prescient way, D’Amato turned to existing drugs to probe the potential utility of antiangiogenic therapeutics. He reasoned that birth defects might reflect exposure to antiangiogenic agents in the first trimester of pregnancy, when the growth of blood vessels is tightly orchestrated to limb and organ development. His search quickly focused in on thalidomide and retinoids, the teratogenic effects of which, he thought, might be linked to the inhibition of angiogenesis. D’Amato further proposed that such agents might have the potential for counteracting the aberrant angiogenesis that typified disease states associated with blindness and cancer (22).
As a former student of mine (JWH), D’Amato contacted me with his idea about these drugs, and I encouraged him to submit a patent with claims covering the use of thalidomide and retinoids in treating diseases of angiogenesis. Shortly thereafter, he joined Folkman’s laboratory at Children’s Hospital and began his remarkable career in the field of angiogenesis research. As a founder and CEO of EntreMed (JWH), Dr. D’Amato introduced me to Dr. Folkman, and I began to collaborate and discuss the clinical and commercial feasibility of developing thalidomide as an antiangiogenic agent. In 1993, we initiated a sponsored research program with Children’s Hospital that supported the discovery and characterization of novel antiangiogenic drug candidates. As part of this relationship, EntreMed licensed thalidomide intellectual property rights from D’Amato and Children’s Hospital.
The potential resurrection of thalidomide as a drug was fraught with problems. Thalidomide had been marketed and widely sold in Europe, in the late 1950s and early 1960s, as a sedative. It had appeared remarkably safe, especially compared to the widely abused barbiturates of the day. In some early studies, as much as a gram of thalidomide was given to normal human volunteers without adverse effect. It was also believed to have anti-emetic effects, prompting its use by women in early pregnancy. Only later was it learned that as little as one tablet of thalidomide taken during the first trimester could be disastrous. Birth defects caused by thalidomide would eventually total more than 10,000 cases worldwide.
Thalidomide was never approved for use in the United States due to the actions, foresight, and instincts of Frances Kelsey of the FDA. Thalidomide was the first drug application to which Kelsey was assigned after joining the agency in 1960. She would later recall, “They gave it to me because they thought it would be an easy one to start on....As it tuned out, it wasn’t all that easy” (23). Even though birth defects had not yet been reported when Kelsey began at the FDA, she was not convinced the drug was adequately safe, and she refused to clear it for sale. Follow-on reports of neuritis strengthened her resolve. Ultimately, of the many thousands of babies injured by thalidomide worldwide, there were only seventeen US born cases, each arising from investigational use or from drug obtained abroad. The thalidomide catastrophe would serve as a strong driver for the Kefauver-Harris Amendments, effected into law in October of 1962, which strengthened the FDA’s protective function in drug experimentation on humans. Drug manufacturers would no longer merely have to show that a drug was safe and properly labeled; they became obligated to indicate drug efficacy.
They were further required to obtain informed consent from their human subjects and to report all adverse reactions to the FDA. Kelsey became head of the FDA’s investigational drug branch, created to evaluate and monitor clinical trials for compliance with these new drug regulations.
In the summer of 2001, EntreMed staff appeared at congressional hearings, along with Geraldine Ferraro (whose multiple myeloma was successfully treated with thalidomide) and others, to encourage further clinical exploration of thalidomide. These hearings, which acknowledged the tragic past consequences of thalidomide use, informed the public and FDA about the therapeutic potential of the drug to combat angiogenesis in disease states. An example of this potential was offered in a review of the 1964 discovery, by Israeli physician Jacob Sheskin, of thalidomide’s usefulness in treating nodular lesions associated with leprosy. These lesions are angiogenic, which in retrospect explains the benefits of thalidomide. In 1991, the therapeutic utility of thalidomide surfaced again, when Gila Kaplin and colleagues at The Rockefeller University found preclinical evidence that the drug could suppress tumor necrosis factor (TNF; also called “cachexin”) (24, 25), which was suspected to play a role in the wasting disease (cachexia) associated with HIV/ AIDS. Kaplin and colleagues submitted a patent for the use of thalidomide to inhibit TNF and to prevent cachexia in the treatment of AIDS patients (US Patent 5385901); no claims were made regarding the treatment of angiogenic diseases such as cancer.
In 1993, EntreMed collaborated with Howard Fine at the NCI to initiate phase 2 studies of thalidomide in patients with brain tumors. These studies produced evidence of tumor shrinkage (26) and led to further exploration of thalidomide as a new therapy for cancer patients. As a result, the patents for the use of thalidomide licensed by EntreMed specifically covered angiogenic cancer and blindness (macular degeneration), and they became a key to the intellectual property for the present use of thalidomide in treating cancer (US Patent 5,593,990, 5629327). In this respect, a critical lesson for drug development concerns the importance of appropriately documenting data and making proper claims to secure patents.
Celgene sought significant intellectual property rights to thalidomide and licensed a broad patent portfolio, including the Rockefeller patent, as well as rights from Pediatric Pharmaceuticals for the FDA-approved treatment of leprosy. Celgene implemented creative steps to promote the tight control of distribution, from doctor to pharmacist to patient, in order to preclude misuse of the drug, particularly among women of child-bearing years. Recognizing the value of the EntreMed patent and license for thalidomide, and with an effort to accelerate the process of making the drug available to patients with cancer such as leukemia, EntreMed licensed its portfolio of clinical data and intellectual property to Celgene, in 1996, in exchange for certain payments and royalties. In 1998, marketing approval for Thalomid (thalidomide) was granted by the FDA to Celgene for the treatment of erythema nodosum leprosum (ENL), a severe and debilitating condition associated with leprosy. In 2006, thalidomide in combination with dexamethasone was launched by Celgene for the treatment of multiple myeloma.
Very promising next-generation thalidomide analogs invented by EntreMed scientists, including 3-amino thalidomide and 6-amino thalidomide, proved very effective in arresting the proliferation of endothelial cells and tumor growth.
Subsequently, Celgene’s scientists discovered that 3- amino thalidomide demonstrated antitumor effects. EntreMed, with the prevailing composition of matter patent and earlier filings, joined forces with Celgene to ensure that the promising analogs were brought forward into clinical practice. Subsequently, the FDA approved Revlimid (lenalidomide in combination with dexamethasone) for the treatment of multiple myeloma patients; it is also under study for the treatment of a number of solid tumors. Today, thalidomide and Revlimid have achieved blockbuster status with combined worldwide annual 2008 revenues over $1.8 billion. This astonishing success shows that even a drug with very significant toxicity can be developed for effective use, provided that the appropriate care, persistence, insight, and creativity are practiced.
Future Directions for Antiangiogenics
Drug resistance is a frequent challenge in developing treatments for cancer. Antiangiogenic drug development is no exception, with data demonstrating that resistance to Avastin occurs within a few months after administration (27, 28). Indeed, over twenty different stimulators of angiogenesis exist, including various forms of VEGFs and FGFs. Blocking one of them with a specific antibody doesn’t block them all, and some may be overexpressed when VEGF is blocked or neutralized. The molecular pharmacology and details of the specific pathways involved in antiangiogenic drug resistance are being increasingly characterized, both advancing the science and suggesting new avenues for improved therapy (31). In general, one could also argue that the more specific the antiangiogenic drug in preventing endothelial cell proliferation (e.g., downstream from earlier events in the angiogenesis cascade), the less likely it is that drug resistance will occur. For example, endostatin blocks endothelial cell proliferation regardless of how they were stimulated (VEGF, bFGF, etc.), and potentially derivatives, congeners, or mechanism-related agents may yet offer a lower likelihood of developing drug resistance (15, 32).
Although antiangiogenic agents have been a powerful addition to therapeutics, there are several reasons why they have in some instances failed or been less effective than expected (Box 1). These areas suggest approaches to improving targeted antiangiogenic-based therapy. Clearly, combining antiangiogenics with other compounds that may have synergistic effects (e.g., known chemotherapeutics) offers promise for the future. One such approach is “designer antibodies” engineered to recognize two different and complimentary drug targets. (33). The recent report from Bostrom and colleagues from Genentech on advancing the science and therapeutic use of an antibody that recognizes with high affinity both VEGF and human epidermal growth factor receptor 2 (HER 2) and demonstrates activity in vitro and in mouse xenograft cancer models may be a promising new development (29). This progress also provides an example that therapeutic advances and commercial success (in this case, of Avastin) can further stimulate additional basic scientific advances and research progress.
Why Are Antiangiogenic Drugs Not More Effective in Treating Cancer?
-
Redundancy of proangiogenic systems.
-
Given a multiplicity mediators of angiogenesis, the blocking of one growth factor or one pathway allows other growth factors or other pathways to mediate angiogenic effects.
-
Highly selective drug such as monoclonal antibodies focused on a single target may have optimal safety and minimal off-target effects but are not sufficient to deal with multiple other proangiogenic factors.
-
-
Starving tumors of nutrients may result in a “survival” rebound of tumor cells, metastasizing to other sites in search of oxygen and nutrients.*
-
Tumors themselves may be a direct or indirect source of both pro- and/or antiangiogenic factors that can affect the primary tumor, metastases, or other cancers. Removing the tumor by surgery or reducing the tumor size with drug may change this balance.
-
Increasing the dose of small-molecule inhibitors of angiogenesis may result in increased toxicity. Off-target side effects limit selectivity and ability to increase dosing.
-
Reducing angiogenesis may not be the sole mechanism of action when the drug is used alone or in combination with other drugs.
-
For example, they may change vascular permeability or improve delivery of other chemotherapy agents.
-
Pretreatment circulating levels of VEGF did not predict response to bevacizumab in CRC.**
-
Many antiangiogenic drugs are best used in combination with other agents.
-
Some Further Lessons Learned
The creative process of “seeing things others have seen and thinking things no one else has previously thought” (Szent-Georgyi) and being on the right track may be only one step in the recipe for translation of scientific discoveries: converting these ideas into safe and cost-effective products that make a difference in patients’ lives is another matter. Folkman appreciated this and repeatedly taught that “it is easier to discover drugs than to develop them” (34).
The history of the development of thalidomide, Avastin, angiostatin, and endostatin is a great example of the ways some drugs succeed and others never are commercialized. It takes a combination of factors, including huge resources, adequate drug bioavailability, useful preclinical and clinical metrics of efficacy, solid intellectual property rights, successful and cost effective manufacturing, and a healthy dose of good timing and luck. Smaller companies always have relative resource constraints, and EntreMed was not able to conduct more than a handful of phase 2 clinical studies with endostatin due to the costs of manufacturing as well as the expenses of clinical trials. On the other hand, Genentech was and is far better resourced (for 2008, >20% of revenue spent on research and development), experienced with protein production and manufacturing, and importantly, able to conduct over sixty clinical trials to establish efficacy and a survival advantage for patients treated with their soon-to-be blockbuster antiangiogenic drug Avastin. The research and development of innovative drugs has a low probability of success but is a high risk and high reward business (35).
Postscript
Judah Folkman died in 2008 at the age of seventy-four while traveling to present a scientific lecture. His impact, however, continues. There are more than 42,000 literature citations noted in PubMed for angiogenesis. Major improvements have occurred in cancer treatment resulting from a focus on targeted therapeutics; the top four selling anticancer drugs today are all targeted agents. Avastin is a blockbuster, with $3 billion in worldwide sales. The nature of the pharmaceuticals business is very dynamic with abundant collaborations, mergers, and acquisitions. Indeed, almost unimaginable technology and business success may not be sufficient to remain independent. In Early 2009, Roche completed its acquisition of Genentech, agreeing to pay $46.8 billion in cash to buy the 44% of Genentech that it did not already own. Now, Genentech will become a wholly-owned member of the Roche Group.
A number of new research avenues and approaches for improved drug treatments have been discovered as a result of a greater understanding of the role of angiogenesis in disease. Robert D’Amato’s work in Folkman’s laboratory demonstrating that thalidomide is an angiogenesis inhibitor provided a mechanism and understanding for this drug’s birth-defect side effects. It also suggested new therapeutic uses for thalidomide in inhibiting abnormal angiogenesis in cancer and other diseases.

Judah Folkman
Drug companies are well-positioned to do the integrated R&D necessary for successful follow-up therapies. Lucentis (ranibizumab), a fragment of the monoclonal antibody bevacizumab, was also researched and developed by Genentech and is an important drug for treating macular degeneration, a major cause of blindness; it was launched in the USA in 2006. There are over twenty VEGF-targeted agents in clinical trials as well as several new small-molecule drugs on the market. Examples of these targets are shown in Table 3. The potential of antiangiogenic drugs has begun to be realized, and there is ongoing research and development on the use of proangiogenic agents in a number of high-need areas such as coronary heart disease and wound healing disorders. The prescription for innovative drug success is a difficult one to fill and sustain (Box 2). The persistence of Folkman, Ferrara, and their colleagues, and their correct judgment in championing the role of angiogenesis in cancer, blindness, and as drug targets, were key factors in advancing and accelerating the tremendous progress achieved and paved the way for additional significant future advances in both research and health care (36–38).
A Prescription for Innovative Drug and Team Success
-
A Culture of Innovation
-
Resources: Money and Talent
-
Time
-
Commitment to Win
-
Persistence
-
Judgment
-
Alignment of Stakeholders: Investors, Management, Scientists

The role of angiogenesis in cancer and the influence of pro- and antiangiogenic factors. Adapted from (38).

VEGF, angiogesis, tumors, and inhibition by Avastin. The biological coordination between tumorigenesis and angiogenesis is mediated by proangiogeneic factors such as VEGF. Antibody strategies have resulted in drugs like Avastin that work to bind VEGF, blocking angiogenesis and thereby interfering with the disease. Blue circles represent VEGF; gold circles in right-hand panel represent metastases and possible exosome release.
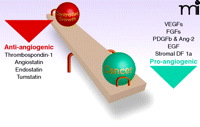
A delicate balance. The “angiogenic switch” includes the results of a balance between positive and negative angiogenesis regulators. Tumor growth is thought ot occur with an imbalance of these factors and the “switch” is turned on in cancer progression.
Endogenous Angiogenic Growth Factors
Endogenous Inhibitors of Angiogenesis
VEGF Pathway Targets: Approaches for Drug Design
Acknowledgments
This perspective is illustrative, subject to the author’s viewpoints and highly condensed. They present key events, but not necessarily all events, contributors or contributions are noted. JWH was a co-founder, CEO and Chairman of EntreMed from 1992–2002.
Footnotes
-
↵* Paez-Ribes, M. et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 15, 220–231 (2009). Ebos, J.M. et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239 (2009).
-
↵** Mass, R.D. et al. Clinical benefit from bevacizumab (BV) in responding (R) and nonresponding (NR) patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol 23, S249–S249 (2005).
- Copyright © 2009
References

John W. Holaday, PhD, is CEO of QRxPharma, a specialty pharmaceutical company focused on pain and CNS diseases. He was co-founder of Medicis Pharmaceutical, and in 1992 he founded EntreMed, where he served as CEO and Chairman until 2002. Dr. Holaday obtained his BS and MS from the University of Alabama and his PhD in pharmacology from the University of California, San Francisco. He served at the Walter Reed Army Institute of Research as an officer and civilian for twenty-one years, and he was Professor of Anesthesiology and Critical Care Medicine at the Johns Hopkins University School of Medicine. Presently, he is adjunct professor of psychiatry at the Uniformed Services University of the Health Sciences. He is on the Advisory Board of Harbert Investments and Hudson Brightwaters investment bank. Dr. Holaday was named to the Ernst & Young Entrepreneur of the Year 2006 Hall of Fame. E-mail john. holaday{at}qrxpharma.com; fax 301-365-8054.

Barry A. Berkowitz, PhD, received his BS in pharmacy from Northeastern University and his PhD in pharmacology from the University of California. He has held senior positions in research at the Roche Institute of Molecular Biology and executive positions in research and development at SmithKline and several biotechnology companies. Dr. Berkowitz has co-founded several companies, including Magainin Pharmaceuticals (Genaera), Strong Pharmaceuticals (Fibrogen), Myco/ChemGenics Pharmaceuticals, and New Chemical Entities. He is an adjunct professor of pharmacology and experimental therapeutics at Boston University School of Medicine and Northeastern University Bouve College of Health Sciences. He is currently CEO of Strong Pharma. E-mail Strongpharma{at}gmail.com; fax 508-877-3286.

