In Search of Analgesia: Emerging Poles of GPCRs in Pain
Abstract
Of all clinically marketed drugs, greater than thirty percent are modulators of G protein–coupled receptors (GPCRs). Nearly 400 GPCRs (i.e., excluding odorant and light receptors) are encoded within the human genome, but only a small fraction of these seven-transmembrane proteins have been identified as drug targets. Chronic pain affects more than one-third of the population, representing a substantial societal burden in use of health care resources and lost productivity. Furthermore, currently available treatments are often inadequate, underscoring the significant need for better therapeutic strategies. The expansion of the identified human GPCR repertoire, coupled with recent insights into the function and structure of GPCRs, offers new opportunities for the development of novel analgesic therapeutics.
Introduction
The G protein–coupled receptors (GPCRs) comprise the largest superfamily of transmembrane receptors. Their function is to transduce extracellular stimuli into intracellular responses. These stimuli can be remarkably diverse, ranging from physical stimuli (e.g., photons or heat) to chemical signals in the form of ions (e.g., Ca2+, H+), chemical neurotransmitters (e.g., dopamine, noradrenaline, adrenaline, acetylcholine, or nucleotides), peptides and protein hormones (e.g., chemokines or opiates), and lipids and eicosanoids (e.g., sphingolipids or leukotrienes). GPCRs mediate and/or modulate virtually all physiological processes in eukaryotic organisms, including acute and chronic pain (1).
Disorders resulting in persistent pain are among the most common forms of chronic illness in North Americans. In individuals age sixty and under, the prevalence of migraine and chronic back pain is ten and fifteen percent, respectively. Arthritis among people less than sixty years of age occurs at a rate of twelve percent, and the frequency rises to forty-six percent for the population that is older than sixty (2). Medical conditions including diabetes, AIDS, and multiple sclerosis all have a high incidence of chronic neuropathic pain. Because pain impairs one’s ability to carry out a productive life, it has serious economic consequences in addition to being a major health problem. In the US alone, an estimated $100 billion is spent each year on health care associated with chronic pain, and an equal amount is further estimated for the related loss of productivity (3, 4). Available therapeutic interventions, such as morphine, are not always able to adequately control pain; not only is drug efficacy at issue, but intolerable side effects, such as sedation, respiratory depression, and gastrointestinal impairment, can also preclude effective pain management. The development of new drugs that target members of the GPCR superfamily holds great promise for the treatment of acute and chronic pain, reaching far beyond the use of traditional opioid receptor agonists.
In this review, we will first provide an overview of GPCR function with regard to the pain signaling system. Second, we will discuss emerging insights into GPCR function that relate to nociceptive transmission. Finally, we will conclude with a brief summary of the role of each GPCR family in nociception.
G Proteins in Signaling
Upon GPCR activation, intracellular signaling systems are activated that couple to a diverse array of downstream effector systems. By definition, signal transduction through GPCRs involves the heterotrimeric GTP-binding proteins (G proteins) to which these receptors are coupled. Current estimates, based on the sequencing of the human genome, predict that the G proteins in human cells can be assembled from among sixteen α, five β, and fourteen γ subunits, with each heterotrimeric combination corresponding to a distinct complement of effector targets (5, 6). The approximately 400 human GPCRs (exclusive of odorant and light receptors) are differentially expressed by specific tissues, allowing for a diversity of signaling cascades that may further be localized with respect to distinct intracellular domains and associated with specific G proteins. In addition, each GPCR may be sensitive to multiple endogenous agonists, and agonists may act at multiple receptor isoforms. Furthermore, data are emerging that GPCRs also elicit G protein–independent intracellular effects, further increasing the spectrum of possibilities (7).
The basic cycle of G protein activation and inactivation is illustrated in Figure 1. Agonist binding and receptor activation induce a conformational change in the heterotrimeric G protein such that the α subunit binds GTP in exchange for GDP, thereby causing the G protein to dissociate into a GTP-bound α monomer and a βγ dimer. The α monomer and βγ dimer are subsequently free to engage target effectors. A mechanism for terminating G–protein signaling to effector systems is built in to the α subunit by means of its intrinsic GTPase activity. Hydrolysis of GTP returns the α subunit to its GDP-bound state, which assembles with the βγ dimer to reform the inactive, heterotrimeric G protein. A number of regulators of G protein signaling, or RGS proteins, enhance the GTPase function of the α subunit and thereby reduce the duration of GPCR signaling. The G protein families primarily involved in the modulation of neurotransmission utilize αs, αi/o, or αq/11 subunits (Figure 1B); members within each family show differences in their patterns of expression (8). Downstream effectors also show isoform-specific intracellular targeting and tissue-specific distribution patterns, providing another level of selectivity in the signaling pathways activated by GPCRs in different cell types.
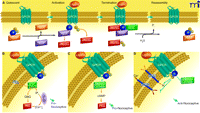
Overview of GPCR signaling. A) Diagram of the cycle of G protein activation and inactivation. In the absence of GPCR signaling (Quiescent), G proteins are present as inactive αβγ trimers; the α subunit is bound to GDP. Binding of ligand to GPCR (Activation) causes a conformational change that promotes binding of the receptor to its preferred trimeric G protein and concomitant displacement of bound GDP by incoming GTP at α subunit. Upon GTP binding, the αβγ trimer dissociates into GTP-bound monomer and βγ dimer, each of which can then interact with respective effectors. Signaling is terminated by the GTPase activity of the α subunit; this GTPase activity can be enhanced by RGS proteins. The α subunit–catalyzed hydrolysis of GTP causes subunits to reassemble into the trimeric G protein. The GPCR is generally desensitized and internalized for recycling or destruction (see text). B–D) Diagrams of the canonical signaling pathways for the major G protein families, as described in the text. (ER, endoplasmic reticulum.)
Gs proteins (i.e., heterotrimeric G proteins that possess an αs subunit) exert their effects primarily by activating adenylyl cyclase, resulting in increased intracellular cyclic AMP (cAMP), which in turn activates downstream effectors, including protein kinase A (PKA). Activated PKA phosphorylates numerous proteins that determine the physiological properties of nociceptors (see below). There are also reports that in some cell types, including some nociceptors (sensory neurons that detect noxious stimuli), Gs signaling can lead to activation of protein kinase C (PKC) through the cAMP-activated guanine exchange factor Epac (9, 10). These phosphorylation events are regulated in turn by phosphatases and their downstream substrates. PKA may also activate transcription factors, including the cAMP response element binding protein (CREB), leading to long-term changes in the physiological properties of affected neurons. Gs activation typically results in increased neuronal excitability.
Gi/o proteins mediate the widespread inhibitory effects of many neurotransmitters. Especially significant for the purposes of our discussion, Gi/o proteins also mediate the effects of almost all analgesic GPCR agonists. Several mechanisms account for the inhibitory activity of Gi/o proteins. First, the GTP-bound αi/o subunit inhibits adenyly cyclase, counteracting the effects of Gs activation. Second, the dimer acts to inhibit voltage-dependent calcium channels, resulting in reduced neurotransmitter release and negative regulation of calcium-activated transcription. Third, they directly hyperpolarize neurons by activation of the G protein–gated inwardly rectifying potassium channels (GIRKs), which results in reduced excitability. In addition to affecting channel activity, Gi/o proteins can also modulate neurotransmitter release by interacting directly with release proteins (11). An important function of αi/o subunits is to activate the ERK/MAPK cascade, resulting in regulation of gene expression. [For a comprehensive review of presynaptic signaling by heterotrimeric G proteins, see (12).]
Gq/11 proteins function mainly through phospholipase C beta (PLCβ), of which there are four known isoforms. PLCβ hydrolyzes membrane phosphatidylinositol-4,5-bisphosphate (PIP2) to form IP3, which evokes release of intracellular calcium stores (by activation of IP3 receptors), and diacylglycerol (DAG); both products lead to activation of protein kinase C (PKC). DAG may also activate protein kinase D. Increased intracellular calcium can promote neurotransmitter release at the presynaptic terminal, activates calmodulin-dependent mechanisms (e.g., calcium/calmodulin-dependent protein kinase), and may lead to transcription factor activation. PKC is a major effector for the functional modulation of neuronal signaling machinery downstream of GPCRs. PKCε appears to be particularly important in the sensitization of primary afferent nociceptors in response to activation of Gq/11 protein–coupled receptors, but other family members contribute to this process as well (13).
Receptor activation can be terminated by G protein–coupled receptor kinases (GRKs) and arrestins [for review, see (14, 15)]. Following prolonged GPCR activation, GRKs phosphorylate the intracellular loops and C terminus of the receptor, which causes arrestins to associate with the GPCR and promotes receptor internalization. Internalized receptors may be recycled or targeted for degradation by ubiquitination. For many receptors, desensitization and internalization appear to be separate processes; the underlying mechanisms are under investigation (14, 15).
In summary, GPCRs alter neuronal functional properties both by covalent modification of the signaling machinery (e.g., phosphorylation) and transcriptional activation of targeted genes. A basic overview of these pathways is provided in Figure 1. Our knowledge of the diverse pathways activated by GPCRs continues to expand [for review see (16)].
Pain Signaling
Normal nociceptive transmission (see Box 1) begins when nociceptive axons innervating the target organ (e.g., skin, viscera, or joint) are activated by noxious stimuli. Primary sensory neurons transmit this information from the periphery to the spinal cord dorsal horn, where the nerve impulse is subject to local modulatory control. A subset of postsynaptic spinal neurons (i.e., secondary sensory neurons) send ascending axons to the thalamus, where they relay the information to higher cortical centers. The ascending fibers also send collateral branches into brainstem (i.e., the rostral ventral medulla) and midbrain regions involved in pain modulation (i.e., the periaqueductal grey) and attention and emotion (i.e., the amygdala). These supraspinal centers in turn send descending projections to the spinal cord that can either inhibit or facilitate nociception. Many of the analgesic medications currently available target GPCRs in these descending pathways (17, 18).
Pain vs Nociception
Pain is defined by the International Association for the Study of Pain as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage.” In contrast, nociception refers to the transduction of noxious stimuli, irrespective of cognitive awareness. A nociceptor is a sensory neuron preferentially sensitive to a noxious stimulus or to a stimulus which would become noxious if prolonged. Excitatory and inhibitory influences on nociception are referred to as pro- or antinociceptive, respectively. Increases and decreases in the experience of pain are referred to as proalgesic and analgesic, respectively.
In chronic pain conditions, the normal regulation of nociceptive signaling may be altered. For example, inflammatory mediators released by peripheral tissues and immune cells in response to injury act at GPCRs to sensitize peripheral nociceptors (see Box 1), making them more responsive to both noxious and innocuous stimuli. Persistent firing of peripheral nociceptors causes spinal cord neurons to become more responsive to nociceptive input through a process known as central sensitization. Sensitization also occurs at higher-order relays in the brain. Neurons at each step in the pain pathway, both ascending and descending, are subject to modulation by GPCRs that thus represent potential targets for therapeutic intervention into persistent pain. An overview of the pain signaling system is provided in Figure 2.
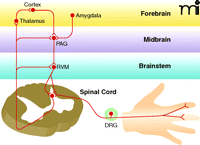
Overview of pain transmission. Nociception begins in the periphery with the activation of nociceptive sensory neurons by noxious stimuli (e.g., heat, acid, or tissue injury). These neurons, which have their cell bodies in the dorsal root ganglia (DRG), synapse on neurons in the spinal cord that send ascending projections to the thalamus, which in turn projects to forebrain regions involved in the subjective experience of pain. Descending inhibitory and excitatory pathways are activated by both ascending input from the spinal cord and descending input from the forebrain and limbic structures, including the amygdala. The major structures modulating descending modulation are found in the brainstem rostral ventral medulla (RVM) and midbrain regions [periaqueductal grey, PAG)].
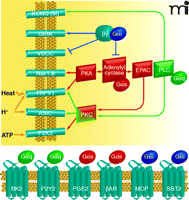
GPCR modulation of nociceptor excitability. GPCR activation, in response to tissue damage or inflammatory mediators, can often result in the covalent modification (e.g., phosphorylation) of ion channels; such channel modification can modulate important physiological properties of nociceptors. A few examples of GPCRs and ion channels that are involved in the modulation of nociceptor activity are shown (see text for details); many other GPCRs have also been implicated in this process. GPCRs also regulate functional properties of neurons at the level of transcription (not shown here). Channels that are regulated by GPCR activation include the ligand-gated ion channels TRPV1, ASICs, and P2X3; voltage-dependent channels including tetrodotoxin-resistant sodium channels Nav1.8 and Nav1.9; voltage-dependent calcium channels; KCNQ channels mediating the M-type potassium current; and G protein–activated potassium channels (GIRK). GPCR-mediated pathways regulating channel function represent an active area of investigation.
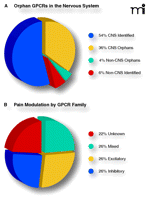
Current status and opportunities in GPCR targeting. A) Predicted proportions of human GPCRs by nervous system expression (90%) and orphan status (40%). Approximately one-third of all GPCR genes encode orphan receptors that are expressed in the CNS. The endogenous ligand(s) and physiological function(s) of these receptors remain to be discovered, representing enormous opportunities for drug development. B) Modulatory effects of GPCRs organized by family. The equal distribution of inhibitory, excitatory, and mixed functional families suggests an equal balance between pro- and anti-nociception.
Emerging Concepts in GPCR Signaling and Pain Modulation
GPCR Signaling and Modulation of Peripheral Nociceptive Channels
GPCRs modulate the function of a wide variety of ion channels and signaling molecules in sensory neurons, allowing neurons to rapidly adjust their sensitivity in response to changes in peripheral target tissues and at the central synapse. In particular, GPCRs modulate ligand-gated and voltage-dependent ion channels that determine key physiological characteristics of nociceptors (19). These channels include members of the transient receptor potential (TRP) family of ligand-gated cation channels, such as TRPV1 and TRPA1, ATP-gated P2X channels, acid-sensing ion channels (ASICs), TTX-resistant sodium channels, voltage-dependent calcium channels, and M-type potassium channels.
TRPV1, a cation channel gated by heat and protons, is selectively expressed in a subset of primary afferent nociceptors and plays a key role in the sensitization of nociceptors in response to inflammation. Numerous GPCRs have been found to regulate TRPV1 (20, 21). Both Gs and Gq protein–coupled receptor signaling enhance TRPV1 function, resulting in peripheral sensitization of nociceptors and reduced pain threshold. Activation of PKCε by Gq signaling plays a major role in the modulation of TRPV1. A related family member, TRPA1, shares many of the same regulatory mechanisms and is largely co-expressed with TRPV1. Several studies suggest that constitutive modulation of TRPV1 by GPCR signaling is required to maintain normal TRPV1 function (20). Additional TRP family members have also been implicated in the transduction of thermal stimuli (21).
P2X3 is a member of the P2X family of ATP-gated ion channels that is preferentially expressed in the non-peptidergic subset of nociceptive sensory neurons. During inflammation, P2X3 currents are enhanced through phosphorylation by PKC. This occurs through an indirect pathway in which the Gs protein–coupled prostaglandin receptor PGE2 acts through Epac1 to activate PKC (9). ASICs are also positively regulated by GPCRs, including serotonin receptors, through the action of PKC. Phosphorylation appears to be selective for the ASIC2b subunit, although it affects currents through heteromeric channels, including those that contain ASIC3 subunits (22). Whereas ASIC2b is expressed by many sensory neurons, ASIC3 is more restricted to peptidergic nociceptors and a subset of larger-diameter neurons of unknown modality (23).
M-type potassium currents, generated by channels consisting of KCNQ subunits, play a key role in regulating nociceptor sensitivity. M-type potassium currents are negatively modulated through Gq protein–mediated signaling; this negative modulation acts to depolarize the resting membrane potential and thereby enhances nociceptive signaling (24). Voltage-dependent calcium channels regulate action potential kinetics and neurotransmitter release as well as neuronal activity–dependent transcription. In nociceptors, these channels contribute significantly to the duration of the action potential. Gi/o protein–coupled receptors such as the opioid receptors inhibit primary afferent signaling in part by inhibiting calcium channels through a direct action of the G protein βγ subunits (25).
Tetrodotoxin-resistant sodium channels Nav1.8 and Nav1.9 are selectively expressed in nociceptors and contribute to injury-evoked changes in neuronal excitability. These channels are modulated through GPCR signaling cascades initiated by inflammatory mediators such as prostaglandins, serotonin, and adenosine (19). Regulation of these sodium channels can occur both through Gs signaling through PKA, which is antagonized by Gi, and by Gq activation of PKC.
Protein Scaffolding and the Organization of Signaling Molecules
The great diversity of GPCR signaling entails a large number of receptors that must evoke selective, often tissue-specific, cellular responses, despite a relatively small complement of G proteins. There is increasing evidence that the functional selectivity of GPCRs is tightly regulated through targeting of signaling components to macromolecular signaling complexes (so-called trans-ducisomes) at specialized membrane compartments known as lipid rafts. Lipid rafts are membrane domains of reduced fluidity, enriched in cholesterol and glycosphingolipids, that promote the assembly of signaling protein complexes (26). These complexes are organized through protein–protein interaction domains (e.g., PDZ, SH2, and SH3 domains) and specialized scaffold proteins that physically coordinate the signaling effector molecules in the transducisome (27). This structural organization allows for highly-efficient regulation of effector function. For example, in some cells, Gq protein–coupled glutamate receptors in the plasma membrane are physically associated with the IP3 receptors on endoplasmic reticulum that regulate calcium stores. This process occurs through binding of the scaffolding protein Homer (28). In nociceptors, some ion channels modulated by G protein signaling are also associated with the transducisome. Examples of channels that are rapidly modulated upon nociceptive GPCR activation and are likely to be associated with scaffolding proteins include the heat- and acid-gated channel TRPV1 and the M-type potassium channel (discussed above) (29).
Available evidence suggests that GPCRs and G proteins are modified by fatty acid acylation, particularly palmitoylation and myristylation, and that these modifications, along with specific polypeptide sequences within the GPCR transmembrane domains, are responsible for directing these proteins to lipid rafts. However, other mechanisms, as yet unidentified, are likely to be important in determining whether a GPCR is targeted to lipid rafts (30). The formation of signaling complexes in lipid rafts provides a mechanism for specialized and highly efficient signal transduction, with pathway selectivity determined by the association of specific effector molecules and receptors through scaffolding proteins.
Integrins are transmembrane proteins associated with lipid rafts and mediate focal adhesions at which the intracellular cytoskeleton connects to extracellular matrix. Integrins contribute to the formation of signaling complexes that are activated in response to binding of extracellular matrix proteins. Studies by Levine and colleagues indicate that integrin binding to the extra-cellular matrix, along with intact lipid rafts, is essential for signaling through a number of GPCRs in inflammatory hyperalgesia (31). The extent to which aberrant nociceptive GPCR signaling or malformation of signaling complexes might underlie persistent pain states is a largely untapped area of investigation.
Ligand-and G Protein–Independent GPCR Signaling
There have been several reports that GPCRs can engage with components of the intracellular signaling complex and activate G protein signaling in the absence of extracellular ligands. Signaling complex components that have been implicated in such interactions include adhesion molecules such as integrins, scaffolding molecules such as Homer, and growth factor receptor tyrosine kinases such as the nerve growth factor receptor TrkA and the epidermal growth factor receptor. This kind of receptor transactivation has been described in both directions; GPCRs may also activate signaling through receptor tyrosine kinases or integrins (16).
In addition, increasing evidence supports the idea that signaling by GPCRs may occur independently of G proteins. The molecules most clearly involved in this process are the β-arrestins, which are widely understood to be involved in the desensitization and recycling of GPCRs. β-arrestins are also able to function as scaffolding molecules for GPCRs and downstream effectors, such as Src tyrosine kinase family members and the MAP kinases, and may actually allow transactivation of these molecules independently of G protein actions (32). However, pathways also exist for the activation of MAP kinases by G protein subunits, suggesting that the pathway used for activation of a specific effector molecule in a given cell type is highly context-dependent (16). These data raise the possibility that there are GPCR-mediated effects in nociceptors that are ligand- and/or G protein–independent (33).
GPCR Oligomerization
It is now recognized that GPCRs, traditionally envisaged monomeric, can form oligomeric complexes. These associations can result in novel pharmacological properties distinct from either component receptor, including alterations in ligand binding affinity, changes in signal transduction, and altered receptor trafficking [for review see (34–37)]. The recognition of oligomeric GCPRs has led to significant re-evaluation of the in vivo mechanisms thought to be involved in GPCR function. Homo- and hetero-oligomerization has been documented within GPCR families, (e.g., the opioid receptor family) and across GPCR families. For example, the functional implications of GPCR–GPCR interactions includes the “unmasking” of opioid binding sites when both the μ- and δ-opioid receptors are co-expressed (38). In addition, the formation of functional GABAB receptors is predicated on a requirement for co-expression of both GABABR1/GABABR2 receptor species (39–41). Although the potential in vivo relevance of these data for neuronal function remains an open question, the existence of oligomers has significant implications for drug development. For example, if the functional receptor is heteromeric, strategies to identify GPCR ligands that rely on cell systems expressing only a single receptor type may not be successful. It is therefore possible that the large number of currently orphaned GPCRs reflects the use of screening paradigms that rely on monomeric rather than heteromeric systems.
Regulation of Cell Surface Expression
The regulation of GPCR internalization and recycling to the cell surface following agonist activation is an area of intense research and has been reviewed extensively elsewhere (42, 43). An interesting new development, with relevance to analgesic drug discovery, is the observation that receptor signaling, internalization, desensitization, and recycling can differ, depending on the specific ligand used (44). This ligand-specific regulation has enormous implications for the development of clinically useful agents with reduced risk of tolerance.
To become functionally competent, GPCRs must be properly synthesized and trafficked to the cell membrane, processes that are under tight cellular control [for review, see (7)]. The cell surface expression of the δ-opioid receptor (DOP) subtype is a case in point. In axon terminals, DOP is associated with large, dense-core vesicles (LDCVs) and in sensory and spinal cord neuron cell bodies, expression is primarily intracellular. In both axons and cell bodies, DOP appears to be inserted into the plasma membrane in a stimulus-dependent manner [see (37, 45, 46)]. DOP may also be translocated in response to chronic morphine exposure, peripheral inflammation, inflammatory mediators, and chronic nociceptive stimuli. As a consequence, sensitivity to DOP agonists is increased. For example, chronic morphine treatment results in an increase both in intrathecal DOP agonist–induced analgesia and in the number of plasma membrane–associated DOP-immunoreactive particles (47).
GPCR Families in Pain Modulation
Early attempts to study, classify, and target GPCRs relied on measurable functional endpoints and on the availability of compounds to selectively stimulate or antagonize those responses. Historically, the modification of these compounds provided the primary approach to the development of new drugs with improved properties. For example, there are at least two dozen different chemical entities in clinical use that target opioid receptors (e.g., morphine and methadone), and most exist in multiple formulations optimized with regard for route of administration or half-life in the plasma. As a result, currently available drugs target only a small fraction of GPCRs.
The human genome project has identified more than 800 different GPCRs, approximately half of which are predicted to respond to endogenous (non-light and non-odorant) ligands (48, 49). Of the 379 GPCRs (i.e., exclusive of odorant and light receptors) by the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification (NC-IUPHAR), an estimated ninety percent are expressed in the central nervous system (50), and nearly forty percent of all GPCRs remain orphans with no identified ligand (49). These numbers predict that over 100 new GPCRs of currently unknown function remain to be identified in the central nervous system, and indeed, new potential targets for drug development have been identified along with previously unknown neurotransmitters. Further insights into GPCR identification and characterization will undoubtedly advance our understanding of pain transmission.
We used the NC-IUPHAR classification system (www.iuphar.org/nciuphar.html) to survey the role(s) of each of the currently proposed GPCR families in pain processing. We were astounded to discover that nearly eighty percent (47/61) of the currently identified families have a known role in the modulation of pain. These survey results speak to the enormous physiological importance of pain modulation by GPCRs. A brief summary of our current understanding of the role of each GPCR family in nociception is provided in Table 1.
GPCR Families and Their G Protein–Dependent Roles in Pro- and Antinociceptive Processing
Conclusions
Regulation of pain transmission by GPCRs occurs throughout the central nervous system, providing a dominant focus for clinical analgesic therapy. However, modulation of nociceptive transduction and processing also occurs in the primary afferent neurons and in peripheral tissues, and all of these sites represent potential targets for novel analgesics. Indeed, it is becoming increasingly clear that GPCRs provide a fundamental mechanism of regulation in an integrated network of communication among sensory axon terminals, their peripheral target tissues, and immune cells (20). The identification of receptors and mechanisms of regulation of GPCRs in pain transmission remains a fertile and largely unexplored field for the development of novel therapeutics for acute and chronic pain, particularly given the paucity of currently available drugs.

Acknowledgments
Acknowledgments
LSS is supported by CIHR MOP-86691 and FRSQ Bourse de chercheur-boursier. DCM is supported by the National Institutes of Health [Grant NS056122].
- Copyright © 2009
References

Laura S. Stone, PhD, received her doctoral degree at the University of Minnesota and postdoctoral training at the Oregon Health and Sciences University (OHSU). Following a brief interlude in biotechnology, she returned to academia and is currently an Assistant Professor in the Faculty of Dentistry at McGill University. Her research program utilizes both human and pre-clinical models to study synergistic interactions among GPCRs in pain and analgesia. Her work also concerns the etiology and treatment of chronic low back pain. Send correspondence to LSS. Email laura.s.stone{at}mcgill.ca; fax 514-398-7203.

Derek C. Molliver, PhD, received his doctoral degree at Washington University in St. Louis and postdoctoral training at the Oregon Health and Science University (OHSU). He is currently an Assistant Professor in the Departments of Medicine and Neurobiology at the University of Pittsburgh and a member of the Pittsburgh Center for Pain Research. His interests are focused on mechanisms underlying the regulation of sensory neuron development and functional plasticity by G protein–coupled receptors and neurotrophic factors.

