High-conductance, Ca2+-activated K+ Channels: Altered Expression Profiles in Aging and Cardiovascular Disease
The high-conductance, Ca2+-activated K+ channel, also designated as the “Maxi-K” or “Big K” (BK) channel, is expressed at high density in the plasma membrane of vascular smooth muscle cells (VSMCs). The opening of these K+ channels exerts a vasodilator influence that is capable of modulating vascular tone and blood pressure. There is strong evidence that reduced numbers or activity of BK channels can trigger anomalous vasoconstriction, disrupt blood flow to critical organs, and elevate blood pressure. This realization has spurred efforts to define the structural components of the channel, characterize its function, and determine its involvement in the accentuated vascular tone associated with aging, metabolic syndrome, diabetes and hypertension.
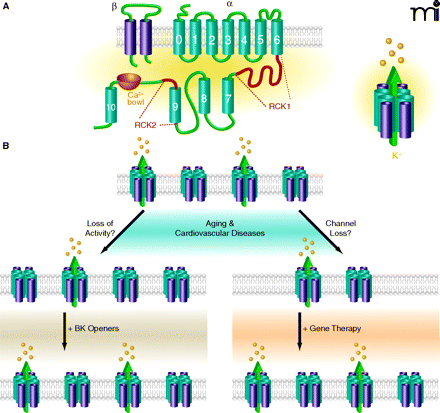
The native BK channel pore is composed of four α-subunits (BKα) that are encoded by a single gene (Slo). Each BKα subunit consists of eleven hydrophobic domains (S0–S10) (Figure 1A). Of the seven (S0–S6) transmembrane domains, S1–S6 share partial sequence similarity with the α subunits of voltage-gated (Kv) channels and contain the pore-forming S5-P-S6 domain and an S4 voltage sensor. An additional S0 domain forms the extracellular N terminus, which couples to the β subunit, permitting interaction. The C-terminal tail contains domains S7–S10 and calcium binding sites (collectively referred to as the “calcium bowl”) as well as the two Regulator of Conductance for K+ domains (RCK1 and RCK2) (1, 2). The heterogeneity of BK channels is conferred by the generation of multiple splice variants and by association of BKα with different β-subunits (BKβ). The BKβ subunits are composed of two trans-membrane domains with a large extracellular loop and cytoplasmic N and C termini (Figure 1A). The predominant BKβ subunit identified in VSMCs is β1, which interacts with the BKα subunit to increase the voltage and calcium sensitivity of the channel.
Functionally, the BK channel participates in the control of vascular tone, promoting vasodilation and buffering vasoconstriction, particularly in small arteries and arterioles that possess high myogenic tone (2). Under resting conditions, the open-state probability of the BK channel is low and its vasodilator influence may be mild. In fact, the association of BKα with the regulatory BKβ1 subunit is required to enhance the Ca2+-sensitivity of the channel and permit basal activity in the physiological range of negative membrane potentials (2). However, in response to excitatory stimuli that trigger arterial activation (i.e., smooth muscle contraction), the ability of the BK channel to oppose vasoconstriction may become profoundly greater, because the depolarization and the rise in cytosolic Ca2+ that occur in VSMCs during vascular activation act synergistically to keep open the BK channel. The resulting K+ efflux causes membrane hyperpolarization, closure of voltage-gated Ca2+ channels, reduced Ca2+ influx, and compensatory vasodilation. Endogenous, vasoactive substances also may influence the activity of the BK channel. These factors include nitric oxide and other endothelium-derived vasodilators, angiotensin II, fatty acid metabolites, steroids, and sex hormones (3).
The BK channel exerts an important vasodilator influence in vivo and contributes to blood pressure regulation. For example, gene deletion of BKα in mice results in a moderate elevation of blood pressure (4). Similarly, mice lacking the critical BKβ1 sub-unit have VSMCs that possess a more depolarized membrane potential, and exhibit excessive vasoconstriction and hypertension compared to wild-type mice (5, 6). Conversely, a gain-of-function mutation in BKβ1 is associated with a low prevalence of human diastolic hypertension (7). It is worth noting that Grimm et al. (8) recently emphasized that the primary ability of the BK channel to suppress blood pressure may not relate to its vasodilator influence but instead to its expression in the renal tubules. Here, the BK channel facilitates K+ secretion in the distal tubule to limit aldosterone release, which in turn, reduces the retention of salt and water. Conversely, the elevation of blood pressure after gene deletion of the BKα or BKβ1 subunit in mice is caused not only by vasoconstriction owing to a loss of BK channel–mediated vasodilation but also by hyperaldosteronism secondary to defective K+ secretion in the kidney (4–6, 8).
Considering the important role of the BK channel in opposing arterial tone, it is not surprising that its potential involvement in cardiovascular disease has drawn attention. There is strong evidence that abnormalities of BK channel expression and regulation contribute to the elevated vascular tone associated with aging, metabolic syndrome, diabetes, and hypertension. During aging, the homeostatic efforts of the vasculature to retain normal vascular tone may be compromised by a reduced number of BK channels and by a loss of the endothelium-derived vasodilator factors that promote channel opening (3, 9, 10). For example, pronounced losses of the BKα and BKβ1 subunits are observed in the coronary arteries of aging F344 rats, and the ability of BK channels to oppose vasoconstriction is compromised in these animals (9, 11). The coronary arteries of elderly human subjects also exhibit a profound loss of BKα and an inability to oppose vasoconstriction (9). In contrast, abnormalities of BK channel expression or dilator function are not evident in the cerebral circulation of aged F344 rats (12). Thus, some vascular beds may be predisposed to elevated vascular tone or vasospasm incurred by a loss of BK channels, whereas others may retain channel expression as a protective mechanism to buffer excessive vasoconstriction. In other cases, mutations may provide a salutary benefit. Senti et al. (13) observed an age-dependent association in women between a gain-of-function BKβ1 mutation that presumably enhanced BK channel activity in vivo and a lower prevalence of diastolic hypertension, myocardial infarction, and stroke.
If aging in humans usually occurs in tandem with cardiovascular diseases, some of which are associated with BK channel abnormalities, what can be said of the animal systems that model cardiovascular disease? Swine models of metabolic syndrome fed an atherogenic diet to induce a clinical profile of hyperlipidemia, impaired glucose tolerance, hyperinsulinemia, and obesity show reduced BK channel-mediated vasodilation and elevated coronary vascular resistance (14). Similarly, small-animal models of metabolic syndrome or diabetes show a loss of BK channel function that relies on the decreased expression of the BKβ1 subunit. This abnormality has been detected in the coronary circulation of Zucker diabetic fatty rats (15) and in the retinal arterioles and cerebral arteries of streptozotocin-induced type-1 diabetic rats and mice, respectively (16, 17). The contribution of BK channels to other common cardiovascular pathologies is not fully resolved, including their role in hypertension, a disease of epidemic proportions. Increased expression of BKα, BKβ1, or both subunits associated with a compensatory vasodilator influence is observed in the vascular beds of genetic and salt-induced rat models of hypertension (18–20). Conversely, a loss of BK channel–mediated vasodilator function, possibly related to the activation of a calcineurin/NFATc3 signaling pathway that decreases BKβ1 expression, is a feature of angiotensin II–induced hypertensive mice (21). Thus, it appears that the behavior of the vascular BK channel during hypertension defies a simplistic explanation, and many factors, including the genetic background, endocrine profile, and age of the experimental animal may influence its expression level and dilator function.
Regardless of its involvement in different pathogenic states, due to its recognized and critical role in mediating vasodilation, the BK channel is regarded as a therapeutic target for the treatment of aging and age-related cardiovascular diseases that are associated with increased vascular tone. Two strategies, not mutually exclusive, have been created to capitalize on the vasodilator action of the BK channel (Figure 1B). First, pharmacological activators have been designed to increase the open-state probability of the channel (3). Notably, this strategy requires that the BK channel is adequately expressed in VSMCs and thus available for drug binding and activation. This scenario may not be present in the VSMCs exposed to aging or cardiovascular pathologies in which a loss of arterialBK channels has been described (9–11, 14–17, 21). At the present time, although some of the BK channel openers are used as research tools to increase channel activity, most clinical trials evaluating BK channel openers as vasodilator therapies have been discontinued, apparently because of the poor potency and lack of selectivity of the compounds (3). Indeed, the therapeutic targeting of the BK channel is confounded by its dense expression in many cell types including neurons, endocrine cells and smooth muscle; only the heart shows a relative deficit of the channel. Nevertheless, some cardiovascular drugs that are in clinical use activate BK channels indirectly by increasing the availability of substances that enhance channel activity. These drugs, including nitric oxide donors, phosphodiesterase inhibitors, and prostaglandin I2 (PGI2) analogs may be particularly helpful in promoting VSMC relaxation when the vascular endothelium is dysfunctional and when nitric oxide and other endogenous endothelium-derived activators of the BK channel are suppressed (3).
As a second strategy to capitalize on the BK channel as a therapeutic target to promote vasodilation, gene therapy is being explored with the intention of restoring or even over-expressing BK channels in diseased VSMCs that may be deficient in one or both of the BK channel subunits (22) (Figure 1B). Although these types of studies are in early-stage trials for the treatment of cardiovascular disease, proof-of-concept that the BK channel can be expressed in smooth muscle to enact relaxation has gained credibility from preclinical and clinical trials designed to correct erectile dysfunction by expressing the BK channel in the corporal cavernosus smooth muscle. In these studies, erectile capability was restored to aged rats and elderly human subjects by the intracavernosal injection of “naked” DNA encoding the human BK channel (23, 24). Additionally, the feasibility of using a viral vector containing a smooth muscle–specific promoter to drive the functional expression of a gene only in VSMCs has been demonstrated by Jiang et al. (25). These investigators used an adenoviral vector containing the promoter of the SM22α gene (which is specifically expressed in smooth muscle cells and not other cell types) to accomplish the preferential expression of a cytochrome P450 enzyme (CYP4A1) in arterial microvessels to mediate angiogenic activity. By using a similar strategy to express therapeutic BK channels selectively in VSMCs, the unwanted side-effects associated with systemic delivery of the BK channel gene to all cell types might be by-passed and offer relief from the lack of selectivity that has limited the use of pharmacological BK channel openers in the past. In summary, if the vasodilator function of the BK channel can be selectively and therapeutically harnessed, it remains a promising drug target to normalize the anomalous vascular tone that is a feature of aging and much cardiovascular pathology.
Li Pang, MD, is an Instructor in the Department of Pharmacology and Toxicology at the University of Arkansas for Medical Sciences. She is particularly interested in the transcriptional and post-transcriptional regulation of Ca2+ and K+ channels and gene therapies to alter ion channel expression in the vasculature. E-mail lpang{at}uams.edu

Nancy J. Rusch, PhD, is Professor and Chair in the Department of Pharmacology and Toxicology at the University of Arkansas for Medical Sciences. Her laboratory focuses on the discovery of vascular Ca2+ and K+ channel abnormalities in hypertension, and identifying target proteins for vasodilator therapies. E-mail nrusch{at}uams.edu
Schematic diagrams of the BK channel structure and the therapeutic strategies to restore BK channel function to the vasculature during aging and cardiovascular diseases. (A) The structure of the pore-forming α subunit (BKα) and regulatory β1-subunit (BKβ1) of the BK channel. Four BKα subunits co-assemble with four BKβ1 subunits to form the channel heteromultimer (shown at right). (B) Schematic diagram depicting BK channel abnormalities in aging and cardiovascular diseases and possible therapies. Left: A disease-related loss of channel activity may be amenable to treatment with BK channel openers that increase channel activity to promote vasodilation. Right: The loss of the BKα and/or BKβ1 channel proteins associated with aging and cardiovascular diseases may be restored by gene therapy using smooth muscle-specific promoters to re-express these subunits as functional BK channels in diseased VSMCs.
- Copyright © 2009

