|
|
Biologie et rôle de CCR5 dans l'infection à VIH et dans son traitement
Michael M. Lederman, MD;
Adam Penn-Nicholson, BA;
Michael Cho, PhD;
Donald Mosier, MD, PhD
RÉSUMÉ
| |
Les récepteurs de chimiokines se trouvent à la surface des
cellules et régulent la migration cellulaire par chimiotaxie. Le CCR5
(CC Chemokine receptor 5) est utilisé par le virus de
l'immunodéficience humaine (VIH) pour infecter les cellules. Des
stratégies ciblant le CCR5 humain sont donc en cours de
développement pour prévenir et traiter l'infection à
VIH. Il peut être envisagé que des stratégies
antirétrovirales ciblant un élément de la cellule
hôte nécessaire à la réplication virale
interfèrent avec sa fonction et puissent ainsi affecter
défavorablement l'hôte. Nous avons mené une revue de
la littérature entre novembre 2005 et avril 2006, en axant notre
recherche sur les articles portant sur la génétique et la
fonction du CCR5, les effets de la délétion de CCR5 dans les
systèmes humain et murin, et les stratégies
thérapeutiques de l'infection à VIH ciblant ce
corécepteur. Les études effectuées chez l'homme et
les murinés, publiées dans la littérature de langue
anglaise entre mars 1996 et avril 2006, ont été
identifiées par une recherche dans MEDLINE en utilisant le terme de
recherche "CCR5". Les articles jugés pertinents, sur la
base de leur titre et de leur résumé, ont été
revus en détails. En outre, en nous basant sur nos connaissances du
domaine et sur autorisation, nous avons également analysé les
travaux non publiés. Dans cet article, nous étudions les effets
potentiels du ciblage de CCR5 sur les défenses des cellules hôtes
chez les individus immunodéprimés infectés par le
VIH.
JAMA. 2006 ; 296: 815-826.
En tant qu'agents pathogènes intracellulaires, les virus
doivent utiliser la machinerie cellulaire de l'hôte pour se
répliquer. En conséquence, le ciblage
d'éléments hôtes nécessaires à la
réplication virale pourrait limiter leur capacité de
réplication.1
Dans d'autres pathologies comme les infections inflammatoires du tube
digestif, la sclérose en plaques, les affections malignes, et les
troubles rhumatologiques, les stratégies ciblant les facteurs
immunitaires de l'hôte comme les cytokines pro-inflammatoires,
2 les
molécules de costimulation,
3 les lymphocytes B,
4 ou les
intégrines, 5
ont prouvé leur efficacité clinique. L'interférence
avec des éléments de défense de la cellule hôte
n'est cependant pas sans conséquences, et est parfois
associée à une augmentation du risque d'infection ou de
néoplasie.6,7
La découverte de l'utilisation de CCR5 (CC Chemokine
receptor 5) par le virus de l'immunodéficience humaine (VIH)
pour pénétrer dans les cellules humaines a conduit au
développement actuel de stratégies ciblant le CCR5 pour
prévenir et traiter l'infection à VIH. Dans ce contexte,
les multiples interactions redondantes entre les chimiokines et leurs
récepteurs permettent une tolérance substantielle de la
délétion ou du blocage de CCR5. Cette tolérance est
reflétée dans la bonne santé généralement
observée chez de nombreuses personnes présentant une
délétion homozygote de 32 paires de bases dans les
séquences codantes pour CCR5 (CCR5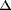 32), qui
rend la protéine dysfonctionnelle. Nous analysons ici la biologie du
CCR5 ainsi que les effets de sa séquestration ou de son inhibition sur
l'immunité de la cellule hôte, et étudions les effets
potentiels de ces stratégies sur les défenses immunitaires
hôtes dans la population générale et chez les personnes
avec une immunodéficience préexistante due à
l'infection à VIH. 32), qui
rend la protéine dysfonctionnelle. Nous analysons ici la biologie du
CCR5 ainsi que les effets de sa séquestration ou de son inhibition sur
l'immunité de la cellule hôte, et étudions les effets
potentiels de ces stratégies sur les défenses immunitaires
hôtes dans la population générale et chez les personnes
avec une immunodéficience préexistante due à
l'infection à VIH.
Nous avons effectué des recherches sur MEDLINE entre novembre 2005
et avril 2006 pour identifier les études humaines et murines de langue
anglaise publiées entre mars 1996 et avril 2006, en utilisant le terme
de recherche "CCR5". Nous avons axé nos recherches sur les
articles portant sur la génétique et la fonction de CCR5, les
effets de la délétion de CCR5 dans les systèmes humain et
murin, et les stratégies thérapeutiques de l'infection
à VIH ciblant ce corécepteur. Les articles jugés
pertinents par leur titre ou leur résumé ont été
revus en détail. Nous avons en outre analysé les recherches non
publiées, en nous basant sur nos connaissances du domaine et
après obtention d'une autorisation.
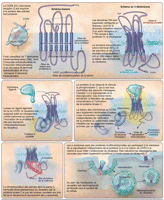
Structure et fonction des récepteurs aux chimiokines
Comme leur nom l'indique, les récepteurs aux chimiokines sont
des récepteurs cellulaires pour les chimiokines – petites
molécules de la famille des cytokines qui induisent la migration
cellulaire par
chimiotaxie.8 Ces
récepteurs ont une structure commune de 7 domaines transmembranaires
(Figure 1),
constituée de boucles extracellulaires et intracellulaires
séparées par des domaines transmembranaires (MSD)
hydrophobiques, caractéristique qui les rend difficiles à
étudier en isolement à moins qu'ils soient exprimés
à la surface d'une cellule. Les récepteurs aux chimiokines
présentent plusieurs autres caractéristiques qui contribuent
à définir leur fonction. Ils sont couplés à des
protéines G et peuvent transmettre des signaux à la cellule en
activant ces protéines ainsi que d'autres molécules signal
(Figure 1).
Lorsqu'ils sont liés par leurs chimiokines, ces récepteurs
peuvent être internalisés, ce qui altère leur
capacité ultérieure de liaison à leurs ligands.
Cependant, une fois internalisés, ils tendent à se recycler
à la surface des cellules à temps. Enfin, de nombreux
récepteurs de chimiokines partagent leurs ligands et vice versa. Ainsi,
un seul chémorécepteur peut se lier et être activé
par plusieurs chimiokines, et une seule chimiokine peut inversement lier et
activer de multiples chémorécepteurs
(Tableau).
|
|
|
|
Figure 1.. Structure et fonction de CCR5 (CC Chemokine receptor 5).
|
|
|
|
|
|
|
Tableau.. Interactions entr les chimiokines et leurs récepteurs*.
|
|
|
Même si l'explication biologique de l'entretien de ces
relations complexes, redondantes et potentiellement inefficaces n'est pas
forcément claire, ces réseaux peuvent être
considérés comme des exemples intéressants du mode de
duplication des mécanismes de défense de l'hôte. Ils
constituent des systèmes de réserve garantissant
l'efficacité des réponses des cellules hôtes aux
provocations microbiennes, ainsi qu'un moyen de surmonter les
contre-attaques microbiennes, de nombreux microorganismes pathogènes
développant des stratégies pour bloquer les chimiokines ou leurs
corécepteurs, ou pour les utiliser dans la
pathogenèse.8
En outre, les chimiokines qui se lient à un même récepteur
peuvent avoir différentes activités biologiques in
vivo, dans la mesure où elles peuvent être exprimées
par des cellules différentes ou sur des sites différents,
où elles peuvent s'exprimer différemment par le biais du
récepteur, et où elles peuvent interagir avec d'autres
récepteurs de chimiokines. Ces relations complexes et redondantes
pourraient expliquer pourquoi la délétion ou le blocage
d'un élément particulier peuvent être
tolérés sans conséquence majeure pour
l'hôte.
Structure et fonction du CCR5
La structure du CCR5 n'a pas encore été résolue,
mais est susceptible de ressembler à celle de la
rhodopsine.9 Les
boucles du récepteur sont probablement disposées en faisceau, de
sorte qu'elles sont plus rapprochées que ce que l'on verrait
dans une configuration linéaire
(Figure 1).
Les chimiokines sont supposées se lier à la partie
amino-terminale de CCR5, puis aux régions des première et
deuxième boucles extracellulaires, bien qu'une analyse
mutationnelle ait indiqué une différenciation potentielle des
chimiokines en matière de sites directs
d'interaction.10
L'extrémité amino-terminale de la chimiokine active la
transmission de signal du récepteur par interaction avec les domaines
transmembranaires groupés du
récepteur.11
La liaison des chimiokines au CCR5 entraîne la dissociation de la
protéine G liée au récepteur, ce qui active la
phospholipase C, qui à son tour induit la formation des seconds
messagers inositol-1,4,5-triphosphate et diacylglycérol; ces
événements conduisent à la libération de calcium
intracellulaire et à l'activation de protéine kinase
C.12,13
Outre les voies activées par ces médiateurs intracellulaires,
des voies indépendantes de la protéine G, telles que celles
médiées par les protéines kinases activées par un
mitogène, pourraient être activées après liaison de
la chimiokine à son récepteur
CCR5.14,15
La phosphorylation des résidus intracytoplasmiques dans le domaine
C-terminal du récepteur active le recrutement de β-arrestines,
protéines multifonctionnelles empêchant un nouveau couplage de la
protéine G, ce qui diminue la transmission de signal par le
récepteur; cela peut également constituer un échafaudage
pour le recrutement d'autres molécules signal. Il est à
noter que les β-arrestines favorisent la liaison du récepteur
à la clathrine pour initier son
internalisation16,17
et son recyclage ultérieur à la surface de la cellule
(Figure 1).
Plusieurs chimiokines peuvent se lier au CCR5 par le biais d'un signal
et promouvoir son internalisation, notamment MIP-1 (macrophage
inflammatory protein-1), MIP-1β, et RANTES (regulated
upon activation normally T-cell expressed and secreted), également
appelées respectivement CCL3 (CC chemokine 3), CCL4 et CCL5
(Tableau). D'autres
chimiokines, notamment MCP-3 (monocyte chemoattractant protein 3),
également appelée CCL7, peuvent se lier au CCR5 en
l'absence de signal et ce faisant, exercer un rôle antagoniste en
interférant avec la liaison d'un ligand (agoniste)
activateur.18 (macrophage
inflammatory protein-1), MIP-1β, et RANTES (regulated
upon activation normally T-cell expressed and secreted), également
appelées respectivement CCL3 (CC chemokine 3), CCL4 et CCL5
(Tableau). D'autres
chimiokines, notamment MCP-3 (monocyte chemoattractant protein 3),
également appelée CCL7, peuvent se lier au CCR5 en
l'absence de signal et ce faisant, exercer un rôle antagoniste en
interférant avec la liaison d'un ligand (agoniste)
activateur.18
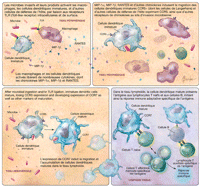
CCR5 et initiation de réponses immunes
De nombreuses cellules de défense hôtes peuvent exprimer le
CCR5 (Encadré), notamment les cellules effectrices de
l'immunité (cellules T, cellules NK et cellules NKT) qui peuvent
produire des cytokines inflammatoires ou détruire les cellules
infectées, et les cellules présentatrices d'antigène
(monocytes, macrophages et cellules dendritiques) qui peuvent amorcer les
réponses immunes. Lorsque les chimiokines se lient au CCR5
exprimé sur les cellules effectrices et les cellules
présentatrices d'antigène, elles peuvent être
activées et induites à migrer. Cependant, parmi ces cellules
exprimant CCR5, seules celles qui co-expriment le CD4 (Encadré)
sont potentiellement susceptibles à l'infection à VIH.
Aux sites d'invasion microbienne, certaines substances communes
à des classes d'agents pathogènes microbiens comme
l'endotoxine, la flagelline, le peptidoglycane, l'ARN à
simple ou double brin, et l'ADN non méthylé contenant des
motifs CpG fréquents dans le génome bactérien, activent
les récepteurs TLR (toll-like receptor) à la surface
des macrophages, des cellules dendritiques, et d`autres cellules, pour initier
la réponse immunitaire innée
(Figure
2).19
À la différence de la grande diversité des
séquences et des structures reconnues spécifiquement par le
système immunitaire adaptatif, un nombre limité de motifs
constituent les signaux activant les TLR. Ainsi, la prolifération
cellulaire n'est pas nécessaire pour générer une
réponse robuste. La réponse innée est donc rapide mais
pas particulièrement spécifique. Elle inclut la production de
cytokines et de chimiokines, qui attirent d'autres cellules de
défense de l'hôte au site d'invasion microbienne. Les
cellules dendritiques cutanées immatures appelées cellules de
Langerhans, qui expriment CCR5, sont attirées vers ces sites par les
fortes concentrations de chimiokines liées au CCR5, exprimées
par les macrophages activés. Les cellules de Langerhans ingèrent
efficacement les microbes et leurs produits de dégradation, et ce
faisant, parviennent rapidement à maturation, en perdant
l'expression de CCR5 et en augmentant l'expression des
molécules de costimulation et de CCR7, chémorécepteur qui
favorise la domiciliation dans le tissu lymphoïde. Dans le tissu
lymphoïde, ces cellules dendritiques matures peuvent présenter les
antigènes étrangers ingérés aux lymphocytes T et B
naïfs pour initier des réponses immunitaires adaptatives plus
spécifiques. Ainsi, l'expression de CCR5 joue un rôle majeur
dans l'initiation des réponses immunitaires adaptatives.
|
|
|
|
Figure 2.. Rôle potentiel de CCR5 (CC Chemokine Receptor 5) dans
l'initiation de la réponse immune.
|
|
|
| Encadré. Leucocytes humains susceptibles d'exprimer
CCR5
- Cellules effectrices de l'immunité
- Lymphocytes T
- Lymphocytes T effecteurs mémoires*
- Lymphocytes T auxiliaires de type 1*
- Lymphocytes T muqueux exprimant*4β7
- Lymphocytes T activés*
- Cellules NK*
- Cellules NKT*
- Cellules présentatrices d'antigène
- Monocytes, macrophages*
- Cellules dendritiques immatures*
- Basophiles
* Peuvent coexprimer le CD4; susceptibles à
l'infection à VIH
Intégrine à la surface des cellules médiant
la domiciliation des cellules immunitaires.
|
|
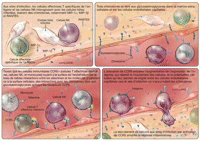
L'expression de CCR5 joue également un rôle dans la
répartition des cellules effectrices aux sites d'infection
microbienne, où elles peuvent participer au contrôle et/ou
à l'élimination des pathogènes
(Figure 3). Aux sites
d'inflammation, les cellules T effectrices spécifiques de
l'antigène et les cellules NK peuvent libérer les
chimiokines MIP-1, MIP-1β, et RANTES – ligands du CCR5- en
se liant aux cellules cibles infectées, afin d'attirer plus de
cellules effectrices vers le
site.19 La
libération de ces chimiokines attire les cellules immunitaires
exprimant CCR5 (cellules T matures avec fonction effectrice, monocytes, et
cellules NK), mais pas les cellules T naïves ou les cellules
mémoire centrales, qui n'expriment pas
CCR5.20,21
Ces chimiokines peuvent se lier aux glycosaminoglycanes, qui sont des
polysaccharides abondamment distribués à la surface des cellules
endothéliales et dans la matrice extracellulaire. Les chimiokines sont
ainsi concentrées sur ces sites et à la surface des cellules
endothéliales
voisines.22 Les
cellules immunitaires circulantes traversent la surface de
l'endothélium en se liant avec une faible affinité aux
sélectines exprimées à la surface cellulaire via leurs
ligands glycoprotéiques de cellules
endothéliales.23
La liaison des chimiokines au CCR5 active les lymphocytes T et la
régulation positive des intégrines β2,
glycoprotéines transmembranaires qui induisent l'adhérence
cellulaire pour arrêter le
roulement.24 La
liaison au CCR5 par une chimiokine induit également la polarisation des
lymphocytes T et leur migration à travers la surface
endothéliale vers le site
inflammatoire.24
Certaines données ont aussi montré que le CCR5 à la
surface des lymphocytes T s'accumule sur la synapse immune, où le
récepteur des lymphocytes T interagit avec le complexe peptide-CMH
(Complexe d'histocompatibilité majeure) sur la cellule
présentatrice d'antigène;
25 cette
interaction pourrait également renforcer l'activation des
lymphocytes T.
|
|
|
|
Figure 3.. Rôle potentiel de CCR5 (CC Chemokine Receptor 5) dans
l'amplification de l'inflammation tissulaire.
|
|
|
CCR5 et infection à VIH
Peu après que le CD4 a été reconnu comme
nécessaire à la réplication du VIH dans les cellules
hôtes, 26 une
série d'expériences a été effectuée
pour déterminer si son expression était suffisante pour
permettre cette réplication. L'expression du CD4 humain sur des
cellules murines n'induisait pas d'infection par le VIH,
27,28
et les expériences dans lesquelles les cellules humaines permissives
étaient fusionnées avec des cellules murines non permissives
indiquaient que l'absence d'entrée du virus
n'était pas liée à un facteur inhibiteur dans les
cellules murines mais à l'absence d'un élément
nécessaire aux premiers événements
postliaison.29 La
chasse au corécepteur du VIH était lancée. En 1995,
Cocchi et coll.30
rapportaient que l'action conjointe des chimiokines MIP-1,
MIP-1β et RANTES pouvait empêcher l'entrée du VIH dans
les cellules hôtes. En l'espace de quelques mois, plusieurs groupes
ont identifié les chémo-récepteurs CXCR4 et CCR5 comme
des corécepteurs clés pour l'entrée du
VIH.31-37
Depuis ces observations, d'autres récepteurs aux chimiokines
susceptibles de favoriser l'infection à VIH in vitro ont
été
identifiés.38
Cependant, CXCR4 et CCR5 se révèlent être les
corécepteurs décisifs dans l'infection à VIH in
vivo. Tandis que la liaison et l'activation de CCR5 peuvent
être effectuées par plusieurs chimiokines, le seul ligand
agoniste défini du CXCR4 est le SDF-1 (stromal-derived
factor-1); l'inactivation de CXCR4 ou de SDF-1 est
létale.8
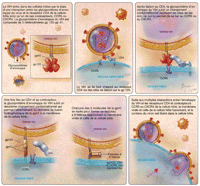
Récepteurs des cellules hôtes et entrée cellulaire du VIH
Comme c'est généralement le cas pour d'autres
agents pathogènes intracellulaires accomplis, le VIH utilise des
éléments de l'hôte hautement préservés
pour entrer dans la cellule, c'est-à-dire le CD4 et le CXCR4 ou le
CCR5. Les mécanismes d'utilisation de ces composants par le VIH
sont progressivement élucidés
(Figure 4). La
protéine d'enveloppe du VIH est constituée de 3
glycoprotéines hétérodimériques, chacune
constituée d'une glycoprotéine 41 transmembranaire
associée de façon non covalente à la glycoprotéine
120. Après liaison de la gp 120 au CD4 cellulaire, la
glycoprotéine d'enveloppe subit un changement conformationnel qui
expose les domaines précédemment inaccessibles, permettant sa
liaison au corécepteur CCR5 ou
CXCR4.34,39,40
Une fois la glycoprotéine d'enveloppe liée au
corécepteur, un autre changement conformationnel majeur est induit dans
le complexe d'enveloppe, découvrant le domaine de fusion
amino-terminal de la gp 41 qui se fixe alors dans la membrane de la cellule
hôte. La membrane virale est ainsi liée à celle de la
cellule hôte. Cette nouvelle conformation permet à chacune des 3
molécules de la gp 41 de se replier pour former un faisceau à 6
hélices rapprochant suffisamment la membrane virale de celle de la
cellule hôte pour promouvoir leur fusion et l'entrée
cellulaire du virus.
|
|
|
|
Figure 4.. Schéma d'entrée du VIH.
|
|
|
Perte ou séquestration de CCR5 et protection contre l'infection à VIH
Peu après l'identification de CCR5 comme corécepteur
clé pour l'entrée cellulaire du VIH, une
délétion de 32 paires de bases a été
identifiée dans le cadre ouvert de lecture du gène CCR5
(région codante) chez quelques individus à risque
élevé d'infection à VIH, mais n'ayant pas
développé la
maladie.41 Il est
important de noter que leurs cellules sanguines étaient
résistantes aux infections par les virus utilisant CCR5 pour leur
entrée cellulaire. Cette délétion de 32 paires de bases
dans le gène CCR5 (CCR532) conduit à la
synthèse d'une protéine tronquée non fonctionnelle,
qui n'est pas exprimée à la surface cellulaire. Fait
surprenant, cet allèle est commun dans la population caucasienne, avec
une prévalence de 10 % à 14
%.42,43
Environ 1 % de la population caucasienne possède 2 copies de ce
gène mutant, et les personnes concernées sont extraordinairement
surreprésentées dans les populations de
séronégatifs à haut risque de
VIH.42,44
L'infection à VIH chez les homozygotes pour CCR532
est extrêmement rare, et lorsqu'elle survient, est causée
par des souches virales utilisant CXCR4 pour pénétrer dans la
cellule.45,46Ainsi,
l'absence congénitale de CCR5 protège contre
l'infection à VIH. Les facteurs limitant l'installation des
infections par les virus utilisant uniquement CXCR4 pour leur entrée
cellulaire ne sont que partiellement connus; ils pourraient refléter
une inhibition partielle de la réplication des virus à tropisme
CXCR4 à des niveaux divers, produisant finalement une barrière
suffisante pour les rendre faiblement
infectieux.47 Les
hétérozygotes porteurs d'un allèle
CCR532 et d'un allèle de phénotype sauvage
peuvent présenter un risque marginalement inférieur
d'infection à VIH,
48,49
et lorsqu'ils contractent l'infection, présentent une
évolution plus
lente.50,51
Des taux inférieurs de charge virale plasmatique peuvent être
observés chez ces
individus.50 Au
cours de l'infection à VIH, environ la moitié des personnes
infectées peut développer des virus susceptibles d'utiliser
le corécepteur CXCR4 pour entrer dans l'organisme (virus
X4).52 Cela est
associé à une accélération de la progression de la
maladie, 53 bien
qu'il ne soit pas clairement déterminé si
l'émergence des virus X4 est la cause ou la conséquence de
la précipitation de l'altération
immunitaire.52
D'autres mutations rares de CCR5 produisant une protéine
dysfonctionnelle ont été identifiées chez certaines
personnes séro-négatives à haut
risque.10,54
En outre, il a été montré que dans différentes
populations, le nombre de duplications du gène MIP-1
prédit inversement le risque d'acquisition du VIH et le taux de
progression de la
maladie.55 Si,
comme on le pense, l'augmentation des duplications du gène
entraîne une plus grande expression du ligand MIP-1 de CCR5,
l'accroissement de l'occupation et de l'internalisation du
récepteur en résultant pourrait limiter la disponibilité
du corécepteur et diminuer le risque d'infection à VIH,
ainsi que la propagation virale. La disponibilité de CCR5 à la
surface cellulaire est donc un facteur déterminant de
susceptibilité à l'infection à VIH et de progression
de la maladie.
L'allèle CCR5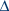 32 et la fonction immunitaire 32 et la fonction immunitaire
Comme nous l'avons évoqué précédemment,
l'allèle CCR532 est fréquent chez les
Caucasiens, mais n'est pratiquement jamais observé dans les
populations asiatiques ou africaines,
43 ce qui indique
qu'il est apparu chez l'homme après divergence de ces
populations. De récentes études ont suggéré
qu'il datait d'au moins 2 900
ans.56
Lorsqu'il est prévalent, il est en équilibre de
Hardy-Weinberg, ce qui suggère qu'au niveau actuel de la
population, il n'y a pas d'avantage ou d'inconvénient
majeur pour le génotype hétérozygote ou homozygote. La
forte pénétrance de cette mutation dans certaines populations
suggère cependant qu'à un moment donné, les
individus porteurs de cet allèle ont dû bénéficier
d'un avantage sélectif majeur. Les candidats à cette
pression de sélection incluent la protection contre la peste
causée par Yersinia pestis, bien que les preuves biologiques
de cet avantage soient pour le moins discutables,
56-58
et la protection contre la variole due au virus Variola parce que le
CCR5 et d'autres chémorécepteurs peuvent être
utilisés par les poxvirus associés pour leur entrée
cellulaire.59 La
distribution géographique restreinte, l'âge estimé de
l'allèle CCR532, et l'apparente
absence du VIH chez l'homme avant le siècle dernier,
60 suggèrent
que le VIH n'a pas eu de rôle dans la sélection pour cet
allèle. Quoiqu'il en soit, les individus homozygotes pour
CCR532 semblent avoir une espérance de vie
normale et ne pas présenter d'altération apparente dans les
risques de troubles infectieux ou immunologiques. La tolérance
apparente de ce phénotype nul (pas d'expression de CCR5) serait
liée à la redondance déjà évoquée
des chimiokines et de leurs ligands. Du fait de ces interactions redondantes,
l'absence d'expression de CCR5 pourrait être compensée
par l'utilisation d'autres chémorécepteurs pour
diriger le trafic et l'activation cellulaires. Cependant, il a
été récemment montré qu'en présence
d'une provocation suffisante, un phénotype clinique est
associé à l'absence congénitale de CCR5 à la
surface des cellules. Ainsi, la survie des allogreffes rénales sans
rejet de l'organe transplanté chez les homozygotes pour
CCR532 est apparemment plus longue que chez les
individus porteurs de l'allèle sauvage,
61 ce qui corrobore
les modèles expérimentaux dans lesquels l'application
d'inhibiteurs de CCR5 prolongeaient la tolérance des allogreffes
chez les
rongeurs.62,63
Plus récemment, une analyse rétrospective effectuée dans
le sud-ouest des États-Unis a révélé que chez les
personnes gravement infectées par le virus West Nile, et plus
particulièrement chez celles ayant une infection mortelle, les
homozygotes pour CCR532 étaient
surreprésentés,
64 ce qui valide
chez l'homme les résultats des études expérimentales
effectuées chez des souris au CCR5
inactivé.65
Dans les systèmes murins, l'inactivation de CCR5 majore
la sévérité de certains processus
infectieux66,67
mais pas d'autres,
68-70
bien que pour certaines infections, la morbidité liée à
l'inflammation puisse être
réduite.71,72
Bien qu'il existe une controverse quant à l'effet
bénéfique potentiel d'un allèle
CCR532 unique sur l'évolution de l'infection
par le virus de l'hépatite C,
73-75
il existe des raisons de penser que les porteurs de CCR532
sont plus susceptibles de résoudre l'infection par le virus de
l'hépatite B (M. Carrington, PhD, communication écrite, 13
avril 2006). Ainsi, la conclusion qui peut provisoirement être
tirée jusqu'à l'obtention de nouvelles données
est que l'homozygotie pour CCR532, qui confère un
haut degré de protection contre l'infection à VIH, est
largement bien tolérée, mais que certaines provocations
d'ampleur suffisante pourraient révéler une
immunodéficience apparente associée à ce génotype.
Il reste à déterminer si d'autres infections, ou
évolutions liées à des infections, sont
gérées plus efficacement par ces hôtes.
Inhibition de l'entrée du VIH dans le traitement de l'infection
Le ciblage de l'entrée cellulaire du virus est une
stratégie prometteuse dans le traitement ou la prévention de
l'infection à VIH. Les agents qui bloquent l'entrée du
VIH peuvent cibler des composants du virus ou de la cellule hôte. Un
nombre limité d'anticorps monoclonaux humains contre la
glycoprotéine d'enveloppe du VIH, capables de neutraliser son
infectiosité et d'empêcher son entrée ont
été développés,
76-79
mais l'administration de plusieurs d'entre eux par voie
intraveineuse n'a pas été associée à un effet
antiviral
durable.80
L'interférence avec la liaison au CD4 et à la gp120 par
administration de CD4 soluble s'est révélée
inefficace dans de premières études,
81,82
mais une protéine de fusion CD4-IgG polyvalente (PRO 542, Progenics
Pharmaceuticals, Tarrytown, New York) a démontré une
activité antirétrovirale in vivo après
administration
intraveineuse.83
Une petite molécule bloquant le site de liaison du CD4 à la
gp120 (BMS 378806, Bristol-Myers Squibb, New York, New York), qui
présente une activité antivirale in vitro,
84 est en cours de
développement dans le cadre d'une stratégie topique de
prévention de la transmission muqueuse du
VIH.85-87
Le premier inhibiteur d'entrée approuvé en usage clinique
est un peptide administré par voie parentérale contenant des
séquences de la gp41 du
VIH.88 Ce peptide,
l'enfuvirtide (Roche/Trimeris, Bâle, Suisse/Durham, Caroline du
Nord, États-Unis), bloque la fixation de la gp 41
(Figure 4) qui permet la
fusion entre les membranes virale et cellulaire.
Inhibition de CCR5 dans le traitement ou la prévention de l'infection à VIH
Sachant que le CCR5 est nécessaire à l'entrée du
VIH dans la cellule et qu'il constitue le corécepteur clé
pour la plupart des souches du virus chez les personnes infectées, le
développement de diverses stratégies préventives et
thérapeutiques ciblant ce composant est actuellement en cours. Il est
important de comprendre que différentes stratégies de blocage de
CCR5 peuvent avoir différents effets sur l'hôte et
différentes interactions avec le VIH. En outre, parallèlement
à la progression de la maladie, une proportion croissante de personnes
abrite des virus réplicatifs capables de se fixer indifféremment
sur CCR5 ou CXCR4 (double tropisme R5X 4) ou utilisant exclusivement CXCR4
(tropisme X4) pour entrer dans la
cellule.89 Les
effets thérapeutiques des combinaisons incluant un inhibiteur de CCR5
n'ont pas été étudiés de manière
exhaustive chez ces individus, mais pourraient être concluants si
d'autres agents thérapeutiques restent actifs contre les souches
X4. Par ailleurs, si la combinaison thérapeutique ne supprime pas
complètement la réplication virale, des virus X4 peuvent
émerger. Dans ces cas, un suivi rapproché est nécessaire
pour déterminer si la réplication constante des souches X4
accélère la progression de la maladie. Un dépistage
initial des virus à tropisme X4 et R5 est nécessaire pour
résoudre ces questions avant toute initiation d'un traitement par
inhibiteurs de CCR5.
Agonistes de CCR5. Comme nous l'avons évoqué
précédemment, la liaison de la chimiokine au CCR5 est un
événement agoniste qui entraîne la transduction du signal
intracellulaire et l'internalisation du corécepteur. Un certain
nombre d'analogues de chimiokine à l'extrémité
amino-terminale modifiée ont été
développés, dont l'action inhibitrice du VIH est
substantiellement supérieure à celle de RANTES à
l'état
natif.90-92
Ces agents se lient au CCR5 et activent son internalisation. Leur puissance
est directement liée à l'ampleur et à la
durée de l'internalisation du récepteur; l'action du
plus puissant, PSC-RANTES (N-
(n-nonanoyl)--des-Ser1-
[L-thioproline2,
L--cyclohexylglycine3]
(PSC) RANTES), peut s'étendre à 24
heures.90 En
promouvant la séquestration intracellulaire du corécepteur, ces
agents ne sont pas susceptibles de favoriser l'émergence
d'isolats résistants au traitement et capables d'utiliser
CCR5 pour entrer dans la cellule. L'application vaginale de la PSC-RANTES
chez le macaque rhésus a conféré une protection de niveau
élevé contre l'infection par SHIV 162P3, un virus
chimérique de l'immunodéficience simienne contenant des
séquences d'enveloppe du
VIH.93 Même
si cette stratégie n'a pas encore été
associée à des survenues d'inflammation locale, cette
possibilité ne doit pas être négligée au cours de
son développement et de celui de produits associés. Bien que la
liaison des ligands au CCR5 active la transmission de signaux et
l'internalisation du récepteur, ces événements
peuvent ne pas être nécessairement liés. En
théorie, des agents liant et internalisant le CCR5 sans activité
agoniste peuvent donc être développés. À titre
d'exemple, nous avons récemment trouvé que la
béta-défensine 3, un petit peptide cationique, peut être
antagoniste de la liaison du ligand à l'autre corécepteur
du VIH, CXCR4, et promouvoir son internalisation sans présenter
d'activité
agoniste.94,95
Antagonistes de CCR5. Il existe 2 classes d'antagonistes de
CCR5 en cours de développement, un anticorps monoclonal anti-CCR5 et
plusieurs petites molécules antagonistes. L'anticorps monoclonal
humanisé HGS Ab004 (Human Genome Sciences, Rockville, Maryland) se lie
à la seconde boucle extracellulaire de CCR5, inhibant ainsi la liaison
à la chimiokine et à l'enveloppe du
VIH.96
L'activité des molécules inhibitrices de CCR5, comme
l'aplaviroc (GlaxoSmithKline, Philadelphie, Pennsylvanie), le maraviroc
(Pfizer, New York, New York) et le vicriviroc (Schering-Plough, Kenilworth,
New Jersey), a été testée dans des essais humains
à grande échelle. Chacune de ces petites molécules est
supposée fonctionner comme un inhibiteur allostérique qui
confine le CCR5 dans une conformation telle qu'il n'est plus capable
de se lier à la protéine d'enveloppe du VIH. Chacune peut
fonctionner en tant qu'antagoniste du récepteur, en bloquant
à des degrés divers les signaux induits par différents
chémorécepteurs.97
Aucun de ces agents n'est supposé activer la transmission de
signaux et l'internalisation des récepteurs, et n'est donc
susceptible de promouvoir l'inflammation comme peuvent le faire les
agonistes. Cependant, dans la mesure où le CCR5 est maintenu à
la surface de la cellule, des mutants résistants toujours capables de
l'utiliser pour entrer dans la cellule peuvent être
sélectionnés.98
Les antagonistes se révèlent capables d'induire une liaison
prolongée au récepteur in vitro et in
vivo.99 En
inhibant l'activité de CCR5, ils peuvent bloquer le trafic et
l'activation cellulaires médiés par CCR5. Ces
molécules ont rencontré quelques difficultés au cours de
leur développement. Les essais sur l'aplaviroc ont
été arrêtés pour cause
d'hépatotoxicité100;
un essai sur le vicriviroc chez des patients naïfs de traitement a
été arrêté pour cause d'échec
thérapeutique,
101 et une
étude de phase II du vicriviroc a été
décodée en raison de la survenue inattendue d'affections
malignes, notamment de
lymphomes.102 Il
est intéressant de noter que chez les patients VIH
hétérozygotes (1 allèle sauvage et 1 allèle
CCR532) pour CCR5, l'incidence de lymphome non
hodgkinien malin se révèle inférieure aux
prévisions.103
La relation entre le traitement par vicriviroc et le développement de
ces affections malignes reste incertaine. Les résultats de nouvelles
études, notamment des études en cours sur le vicriviroc et le
maraviroc, sont nécessaires pour déterminer
l'efficacité et l'innocuité des antagonistes de CCR5
dans le traitement de l'infection à VIH.
Inhibiteurs de CCR5 non agonistes non antagonistes
Pro-140 est un anticorps monoclonal humanisé anti-CCR5 (Progenics
Pharmaceuticals) qui inhibe l'entrée du VIH en se liant à
la deuxième boucle extracellulaire de CCR5, mais n'active ni ne
bloque la fonction du récepteur à des niveaux suffisants pour
empêcher l'entrée du
virus.104 Cette
activité sélective surprenante pourrait également rendre
cet inhibiteur moins immunosuppresseur que les inhibiteurs antagonistes.
Conséquences potentielles de l'inhibition de CCR5
Comme nous l'avons évoqué précédemment et
selon la stratégie appliquée, le blocage de CCR5 peut induire
des réponses inflammatoires par activation du récepteur ou
bloquer par antagonisme le trafic et l'activation cellulaires
médiées par CCR5. Si la compensation de l'absence
congénitale de CCR5 par un réseau redondant d'interactions
chimiokine-ligand peut être raisonnablement bien tolérée
à moins d'être fortement provoqué, il n'est pas
clairement déterminé si de graves effets résultant
d'un blocage pharmacologique ou immunologique de CCR5 seraient aussi bien
tolérés. En outre, dans le cadre de
l'immunodéficience liée à l'infection à
VIH, il n'est pas clairement déterminé si l'inhibition
de CCR5 serait aussi compensée. Plusieurs hypothèses explorant
les conséquences potentielles d'une stratégie
thérapeutique inhibant CCR5 dans l'infection à VIH sont
évoquées ci-après.
L'inhibition de CCR5 bloquera-t-elle le trafic des cellules
effectrices vers les sites tissulaires d'inflammation? Cela constitue une
conséquence plausible de cette inhibition et pourrait être
sous-jacent à la tolérance renforcée de l'allogreffe
ainsi qu'à la majoration apparente du risque d'infection
grave par le virus West Nile chez les homozygotes pour CCR532.
Cet effet pourrait même être plus prononcé chez les
individus n'ayant pas connu des années d'accommodation
à l'absence de ce récepteur ou chez ceux présentant
une immunodéficience liée au VIH. Afin de tester cette
hypothèse, l'influx de cellules effectrices dans les sites
tissulaires d'inflammation pourrait être étudié par
analyse immunohistochimique de l'infiltration cellulaire à des
sites biopsiés pour déterminer la position de
l'antigène dans des tests cutanés, chez des personnes
traitées par antagonistes de CCR5 et chez des témoins.
Une minorité d'individus à un stade avancé de
l'infection à VIH présente un syndrome inflammatoire de
restauration immunitaire (IRIS) après administration de traitements
antirétroviraux
suppresseurs.105,106
Ce syndrome inflammatoire morbide a été attribué à
l'entrée brutale des lymphocytes effecteurs aux sites
d'infection opportuniste non reconnue. Si c'est le cas, on peut
supposer que l'incidence d'IRIS serait inférieure chez les
patients traités par une combinaison thérapeutique incluant un
antagoniste de CCR5.
Le blocage de CCR5 affectera-t-il la reconstitution du tissu lymphoïde
associé au tube digestif? Les études chez l'homme et le
macaque rhésus indiquent que les lymphocytes CD4 du GALT sont
rapidement et profondément réduits dans la phase aiguë des
infections à VIH ou à
SIV.107-110
Ces cellules cibles sont presque toutes CCR5 +. Bien qu'il existe une
controverse quant à savoir si cette population cellulaire est
restaurée avec les traitements antirétroviraux, il peut
être supposé que l'administration d'inhibiteurs de CCR5
puisse bloquer ou retarder cette restauration si le recrutement des cellules
sur ces sites nécessite des CCR5 fonctionnels.
Le blocage de CCR5 atténuera-t-il la réponse à
l'immunisation? Ce point constitue également une
conséquence plausible de l'inhibition de ce récepteur, dans
la mesure où le rassemblement des cellules présentatrices
d'antigène aux sites de confrontation antigénique est
supposé médié, au moins en partie, par l'activation
de CCR5. Cela peut être évalué par l'étude des
réponses des lymphocytes T et B à l'administration
vaccinale111,112
chez des personnes traitées par combinaisons antirétrovirales
incluant des inhibiteurs de CCR5 et chez des témoins
appariés.
L'administration d'inhibiteurs de CCR5 augmentera-t-elle le
risque d'infections opportunistes et de néoplasies chez les
patients VIH? Cette question constitue bien sûr un problème de
sécurité fondamental pour cette nouvelle classe
d'antirétroviraux. Alors que l'absence congénitale de
CCR5 est généralement bien tolérée, il se confirme
progressivement que face à une provocation suffisante, les personnes
ayant ce génotype peuvent présenter une altération de la
réponse immune. La manière dont cela se traduira chez les
personnes n'ayant pas bénéficié de
l'accommodation de toute une vie à l'absence de ce
récepteur et susceptibles de présenter des degrés
variables d'immunodéficience liée au VIH reste à
déterminer. Un suivi clinique et immunologique rapproché des
essais cliniques sur les inhibiteurs de CCR5 est donc justifié. Cette
classe d'antirétroviraux potentiellement prometteuse est
également tout à fait capable de représenter un subtil
compromis immunitaire. La manière dont les activités de ces
molécules s'équilibrent chez les individus aux
différents stades de l'infection à VIH et à
différents niveaux de résistance
antirétrovirale113
déterminera leur place dans le traitement de l'infection.
En conclusion, le chémorécepteur CCR5 contribue, par
l'intermédiaire d'interactions avec ses ligands, à
initier les réponses immunes et à répartir les cellules
immunitaires effectrices aux sites d'inflammation. Le CCR5 est
également reconnu comme un récepteur cellulaire clé,
nécessaire dans la quasi-totalité des cas d'infection
à VIH. Des stratégies préventives et
thérapeutiques ciblant ce récepteur sont donc en cours de
développement. Bien que la délétion de CCR5 soit
généralement bien tolérée chez la souris et chez
l'homme, une réponse immune altérée de
l'hôte peut être démontrée dans les deux cas
avec une provocation suffisante. Il reste à déterminer si
l'inhibition pharmacologique de la fonction de CCR5 sera également
tolérée ou si le blocage de ce récepteur aura des effets
défavorables particuliers chez les individus présentant une
immunodéficience sousjacente liée au VIH.
Informations sur les auteurs
Correspondance: Michael M. Lederman, MD, Division of Infectious
Diseases, Case Western Reserve University, 2061 Cornell Rd, Cleveland, OH
44106
(MXL6{at}case.edu).
Contributions des auteurs: le Dr Lederman a eu un accès
complet à toutes les données de l'étude et accepte
la responsabilité de l'intégrité des données
et de l'exactitude de l'analyse des données.
Conception et schéma de l'étude: Lederman,
Penn-Nicholson, Cho.
Recueil des données: Lederman.
Analyse et interprétation des données: Lederman,
Mosier.
Rédaction du manuscrit: Lederman, Cho, Mosier.
Revue critique du manuscrit: Lederman, Penn-Nicholson, Cho,
Mosier.
Obtention du financement: Lederman.
Aide administrative, technique ou matérielle: Cho.
Supervision de l'étude: Lederman, Mosier.
Liens financiers: le Dr Lederman a déclaré qu'il
avait été consultant rétribué par Roche-Trimeris
et avait bénéficié de contrats pour des essais cliniques
de la part de Roche-Trimeris, Schering-Plough, et de Human Genome
Sciences.
Financement/Soutien: ce travail a bénéficié du
soutien sous la forme de bourses de recherche AI 51649, AI 36219, AI 25879 du
National Institutes of Health et du James B. Pendleton Charitable Trust ainsi
que de La Jolla Foundation for Microbicide Research.
Rôle du sponsor: les agences ayant financé n'ont
joué aucun rôle dans la préparation, la revue ou
l'approbation de cet article.
Présentation antérieure: présenté
partiellement au cours de l'atelier HIV Entry Inhibitors Workshop,
Bethesda, Maryland, 2 décembre 2005.
Affiliations des auteurs : Center for AIDS Research, Case Western Reserve University School of Medicine, University Hospitals of Cleveland, Cleveland, Ohio
BIBLIOGRAPHIE
| |
1. Lederman MM. Host-directed and immunebased therapies for humanimmunodeficiency virus infection. Ann Intern Med.1995; 122:218-222.
FREE FULL TEXT
2. Nash PT, Florin TH. Tumour necrosis factor inhibitors.Med J Aust. 2005;183: 205-208.
PUBMED
3. Ruderman EM, Pope RM. The evolving clinical profile of abatacept(CTLA4-Ig): a novel co-stimulatory modulator for the treatment of rheumatoidarthritis. Arthritis Res Ther. 2005;7 (suppl 2):S21-S25.
PUBMED
4. Hasler P. Biological therapies directed against cells in autoimmunedisease. Springer Semin Immunopathol.2006; 27:443-456.
PUBMED
5. Miller DH, Khan OA, Sheremata WA, et al. A controlled trial ofnatalizumab for relapsing multiple sclerosis. N Engl JMed. 2003; 348:15-23.
PUBMED
6. Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, MontoriV. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of seriousinfections and malignancies: systematic review and metaanalysis of rareharmful effects in randomized controlled trials. JAMA.2006; 295:2275-2285.
FREE FULL TEXT
7. Yousry TA, Major EO, Ryschkewitsch C, et al. Evaluation of patientstreated with natalizumab for progressive multifocal leukoencephalopathy.N Engl J Med. 2006;354: 924-933.
PUBMED
8. Murphy PM. Chemokines. In: Paul WE, ed. FundamentalImmunology. 5th ed. Philadelphia, Pa: Lippincott Williams &Wilkins; 2003: 801-840.9. Palczewski K, Kumasaka T, Hori T, et al. Crystal structure ofrhodopsin: a G protein-coupled receptor. Science.2000; 289:739-745.
FREE FULL TEXT
10. Howard OM, Shirakawa AK, Turpin JA, et al. Naturally occurring CCR5extracellular and transmembrane domain variants affect HIV-1 co-receptor andligand binding function. J Biol Chem.1999; 274:16228-16234.
FREE FULL TEXT
11. Oppermann M. Chemokine receptor CCR5: insights into structure,function, and regulation. Cell Signal.2004; 16:1201-1210.
PUBMED
12. Aramori I, Ferguson SS, Bieniasz PD, Zhang J, Cullen B, Cullen MG.Molecular mechanism of desensitization of the chemokine receptor CCR-5:receptor signaling and internalization are dissociable from its role as anHIV-1 co-receptor. EMBO J. 1997;16: 4606-4616.
PUBMED
13. Zhao J, Ma L, Wu YL, Wang P, Hu W, Pei G. Chemokine receptor CCR5functionally couples to inhibitory G proteins and undergoes desensitization.J Cell Biochem. 1998;71: 36-45.
PUBMED
14. Dairaghi DJ, Franz-Bacon K, Callas E, et al. Macrophageinflammatory protein-1 induces migration and activation of human thymocytes.Blood. 1998; 91:2905-2913.
FREE FULL TEXT
15. Del Corno M, Liu QH, Schols D, et al. HIV-1 gp120 and chemokineactivation of Pyk2 and mitogenactivated protein kinases in primary macrophagesmediated by calcium-dependent, pertussis toxininsensitive chemokine receptorsignaling. Blood. 2001;98: 2909-2916.
FREE FULL TEXT
16. Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by-arrestins. Science. 2005;308: 512-517.
FREE FULL TEXT
17. Mueller A, Strange PG. The chemokine receptor, CCR5. IntJ Biochem Cell Biol. 2004;36: 35-38.
18. Blanpain C, Migeotte I, Lee B, et al. CCR5 binds multipleCC-chemokines: MCP-3 acts as a natural antagonist.Blood. 1999; 94:1899-1905.
FREE FULL TEXT
19. Luster AD. The role of chemokines in linking innate and adaptiveimmunity. Curr Opin Immunol. 2002;14: 129-135.
PUBMED
20. Luther SA, Cyster JG. Chemokines as regulators of T celldifferentiation. Nat Immunol. 2001;2: 102-107.
PUBMED
21. Nieto M, Navarro F, Perez-Villar JJ, et al. Roles of chemokines andreceptor polarization in NKtarget cell interactions. JImmunol. 1998; 161:3330-3339.
FREE FULL TEXT
22. Handel TM, Johnson Z, Crown SE, Lau EK, Proudfoot AE. Regulation ofprotein function by glycosaminoglycans – as exemplified by chemokines.Annu Rev Biochem. 2005;74: 385-410.
PUBMED
23. Vestweber D, Blanks JE. Mechanisms that regulate the function ofthe selectins and their ligands. Physiol Rev.1999; 79:181-213.
FREE FULL TEXT
24. Wong MM, Fish EN. Chemokines: attractive mediators of the immuneresponse. Semin Immunol. 2003;15: 5-14.
PUBMED
25. Molon B, Gri G, Bettella M, et al. T cell costimulation bychemokine receptors. Nat Immunol. 2005;6: 465-471.
PUBMED
26. Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF,Weiss RA. The CD4 (T4) antigen is an essential component of the receptor forthe AIDS retrovirus. Nature. 1984;312: 763-767.
PUBMED
27. Jasin M, Page KA, Littman DR. Glycosylphosphatidylinositol-anchoredCD4/Thy-1 chimeric molecules serve as human immunodeficiency virus receptorsin human, but not mouse, cells and are modulated by gangliosides. JVirol. 1991; 65:440-444.
FREE FULL TEXT
28. Tersmette M, van Dongen JJ, Clapham PR, et al. Humanimmunodeficiency virus infection studied in CD4-expressing human-murine T-cellhybrids. Virology. 1989;168: 267-273.
PUBMED
29. Weiner DB, Huebner K, WilliamsWV, Greene MI. Human genes other thanCD4 facilitate HIV-1 infection of murine cells.Pathobiology. 1991;59: 361-371.
PUBMED
30. Cocchi F, DeVico AL, Garzino DA, Arya SK, Gallo RC, Lusso P.Identification of RANTES, MIP-1, and MIP-1 as the major HIV-suppressivefactors produced by CD8 T cells. Science.1995; 270:1811-1815.
FREE FULL TEXT
31. Alkhatib G, Combadiere C, Broder CC, et al. CC CKR5: a RANTES,MIP-1, MIP-1 receptor as a fusion cofactor for macrophage-tropic HIV-1.Science. 1996;272: 1955-1958.
FREE FULL TEXT
32. Arenzana-Seisdedos F, Virelizier JL, Rousset D, et al. HIV blockedby chemokine antagonist. Nature. 1996;383: 400.
PUBMED
33. Bleul CC, Farzan M, Choe H, et al. The lymphocyte chemoattractantSDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry.Nature. 1996; 382:829-833.
PUBMED
34. Choe H, Farzan M, Sun Y, et al. The -chemokine receptors CCR3 andCCR5 facilitate infection by primary HIV-1 isolates.Cell. 1996; 85:1135-1148.
PUBMED
35. Deng H, Liu R, Ellmeier W, et al. Identification of a majorco-receptor for primary isolates of HIV-1. Nature.1996; 381:661-666.
PUBMED
36. Dragic T, Litwin V, Allaway GP, et al. HIV-1 entry into CD4 cellsis mediated by the chemokine receptor CC-CKR-5 Nature.1996; 381:667-673.
PUBMED
37. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor:functional cDNA cloning of a seventransmembrane, G protein-coupled receptor.Science. 1996;272: 872-877.
ABSTRACT
38. Unutmaz D, KewalRamani VN, Littman DR.Gprotein-coupled receptors inHIV and SIV entry: new perspectives on lentivirus-host interactions and on theutility of animal models. Semin Immunol.1998; 10:225-236.
PUBMED
39. Trkola A, Dragic T, Arthos J, et al. CD4-dependent,antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5.Nature. 1996; 384:184-187.
PUBMED
40. Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, HendricksonWA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4receptor and a neutralizing human antibody. Nature.1998; 393:648-659.
PUBMED
41. Liu R, Paxton WA, Choe S, et al. Homozygous defect in HIV-1coreceptor accounts for resistance of some multiply-exposed individuals toHIV-1 infection. Cell. 1996;86: 367-377.
PUBMED
42. Dean M, Carrington M, Winkler C, et al. Genetic restriction ofHIV-1 infection and progression to AIDS by a deletion allele of the CKR5structural gene: Hemophilia Growth and Development Study, Multicenter AIDSCohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort,ALIVE Study. Science. 1996;273: 1856-1862.
FREE FULL TEXT
43. Stephens JC, Reich DE, Goldstein DB, et al. Dating the origin ofthe CCR5-32 AIDS-resistance allele by the coalescence of haplotypes.Am J Hum Genet. 1998;62: 1507-1515.
PUBMED
44. Salkowitz JR, Purvis SF, Meyerson H, et al. Characterization ofhigh-risk HIV-1 seronegative hemophiliacs. ClinImmunol. 2001; 98:200-211.
45. Gorry PR, Zhang C, Wu S, et al. Persistence of dual-tropic HIV-1 inan individual homozygous for the CCR5 32 allele.Lancet. 2002; 359:1832-1834.
PUBMED
46. Michael NL, Nelson JA, Kewal Ramani VN, et al. Exclusive andpersistent use of the entry coreceptor CXCR4 by human immunodeficiency virustype 1 from a subject homozygous for CCR5 32. J Virol.1998; 72:6040-6047.
FREE FULL TEXT
47. Margolis L, Shattock R. Selective transmission of CCR5-utilizingHIV-1: the'gatekeeper'problem resolved? Nat RevMicrobiol. 2006; 4:312-317.
48. Samson M, Libert F, Doranz BJ, et al. Resistance to HIV-1 infectionin caucasian individuals bearing mutant alleles of the CCR-5 chemokinereceptor gene. Nature. 1996;382: 722-725.
PUBMED
49. Marmor M, Sheppard HW, Donnell D, et al. Homozygous andheterozygous CCR5-32 genotypes are associated with resistance to HIVinfection. J Acquir Immune Defic Syndr.2001; 27:472-481.
PUBMED
50. Ioannidis JP, Rosenberg PS, Goedert JJ, et al. Effects of CCR5-32,CCR2-64I, and SDF-13A alleles on HIV-1 disease progression: an internationalmetaanalysis of individual-patient data. Ann InternMed. 2001; 135:782-795.
51. Zimmerman PA, Buckler-White A, Alkhatib G, et al. Inheritedresistance to HIV-1 conferred by an inactivating mutation in CC chemokinereceptor 5: studies in populations with contrasting clinical phenotypes,defined racial background, and quantified risk. MolMed. 1997; 3:23-36.
PUBMED
52. Koot M, Keet IP, Vos AH, et al. Prognostic value of HIV-1syncytium-inducing phenotype for rate of CD4cell depletion and progression toAIDS. Ann Intern Med. 1993;118: 681-688.
FREE FULL TEXT
53. Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change incoreceptor use coreceptor use correlates with disease progression in HIV-1– infected individuals. J Exp Med.1997; 185:621-628.
FREE FULL TEXT
54. Quillent C, Oberlin E, Braun J, et al. HIV-1-resistance phenotypeconferred by combination of two separate inherited mutations of CCR5 gene.Lancet. 1998; 351:14-18.
PUBMED
55. Gonzalez E, Kulkarni H, Bolivar H, et al. The influence of CCL3L1gene-containing segmental duplications on HIV-1/AIDS susceptibility.Science. 2005;307: 1434-1440.
FREE FULL TEXT
56. Hummel S, Schmidt D, Kremeyer B, Herrmann B, Oppermann M. Detectionof the CCR5-32 HIV resistance gene in Bronze Age skeletons. GenesImmun. 2005; 6:371-374.
57. Elvin SJ, Williamson ED, Scott JC, et al. Evolutionary genetics:Ambiguous role of CCR5 in Y. pestis infection. Nature.2004; 430:417.
PUBMED
58. Mecsas J, Franklin G, Kuziel WA, Brubaker RR, Falkow S, Mosier DE.Evolutionary genetics: CCR5 mutation and plague protection.Nature. 2004; 427:606.
PUBMED
59. Lalani AS, Masters J, Zeng W, et al. Use of chemokine receptors bypoxviruses. Science. 1999;286: 1968-1971.
FREE FULL TEXT
60. Korber B, Muldoon M, Theiler J, et al. Timing the ancestor of theHIV-1 pandemic strains. Science. 2000;288: 1789-1796.
FREE FULL TEXT
61. Fischereder M, Luckow B, Hocher B, et al. CC chemokine receptor 5and renal-transplant survival. Lancet.2001;357:1758-1761.
PUBMED
62. Akashi S, Sho M, Kashizuka H, et al. A novel small-moleculecompound targeting CCR5 and CXCR3 prevents acute and chronic allograftrejection. Transplantation.2005;80:378-384.
PUBMED
63. Grone HJ, Weber C, Weber KS, et al. Met-RANTES reduces vascular andtubular damage during acute renal transplant rejection: blocking monocytearrest and recruitment. FASEB J.1999;13:1371-1383.
FREE FULL TEXT
64. Glass WG, McDermott DH, Lim JK, et al. CCR5 deficiency increasesrisk of symptomatic West Nile virus infection. J ExpMed. 2006;203:35-40.
FREE FULL TEXT
65. Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM.Chemokine receptor CCR5 promotes leukocyte trafficking to the brain andsurvival in West Nile virus infection. J Exp Med.2005;202:1087-1098.
FREE FULL TEXT
66. Ank N, Petersen K, Malmgaard L, Mogensen SC, Paludan SR.Age-dependent role for CCR5 in antiviral host defense against herpes simplexvirus type 2. J Virol.2005;79:9831-9841.
FREE FULL TEXT
67. Machado FS, Koyama NS, Carregaro V, et al. CCR5 plays a criticalrole in the development of myocarditis and host protection in mice infectedwith Trypanosoma cruzi. J Infect Dis.2005;191:627-636.
FREE FULL TEXT
68. Algood HM, Flynn JL. CCR5-deficient mice control Mycobacteriumtuberculosis infection despite increased pulmonary lymphocytic infiltration.J Immunol.2004;173:3287-3296.
FREE FULL TEXT
69. Belnoue E, Kayibanda M, Deschemin JC, et al. CCR5 deficiencydecreases susceptibility to experimental cerebral malaria.Blood. 2003;101:4253-4259.
FREE FULL TEXT
70. Nansen A, Christensen JP, Andreasen SO, Bartholdy C, ChristensenJE, Thomsen AR. The role of CC chemokine receptor 5 in antiviral immunity.Blood. 2002;99:1237-1245.
FREE FULL TEXT
71. Glass WG, Liu MT, Kuziel WA, Lane TE. Reduced macrophageinfiltration and demyelination in mice lacking the chemokine receptor CCR5following infection with a neurotropic coronavirus.Virology.2001;288:8-17.
PUBMED
72. Barr EL, Ouburg S, Igietseme JU, et al. Host inflammatory responseand development of complications of Chlamydia trachomatis genital infection inCCR5-deficient mice and subfertile women with the CCR532 gene deletion.J Microbiol Immunol Infect.2005;38:244-254.
PUBMED
73. Promrat K, McDermott DH, Gonzalez CM, et al. Associations ofchemokine system polymorphisms with clinical outcomes and treatment responsesof chronic hepatitis C. Gastroenterology.2003;124:352-360.
PUBMED
74. Goulding C, McManus R, Murphy A, et al. The CCR5-32 mutation:impact on disease outcome in individuals with hepatitis C infection from asingle source. Gut.2005;54:1157-1161.
FREE FULL TEXT
75. Ahlenstiel G, Woitas RP, Rockstroh J, Spengler U. CC-chemokinereceptor 5 (CCR5) in hepatitis C: at the cross-roads of the antiviral immuneresponse? J Antimicrob Chemother.2004;53:895-898.
FREE FULL TEXT
76. Muster T, Steindl F, Purtscher M, et al. A conserved neutralizingepitope on gp41 of human immunodeficiency virus type 1. JVirol. 1993;67:6642-6647.
FREE FULL TEXT
77. Roben P, Moore JP, Thali M, Sodroski J, Barbas CF III, Burton DR.Recognition properties of a panel of human recombinant Fab fragments to theCD4 binding site of gp120 that show differing abilities to neutralize humanimmunodeficiency virus type 1. J Virol.1994;68:4821-4828.
FREE FULL TEXT
78. Trkola A, Ketas T, Kewalramani VN, et al. Neutralizationsensitivity of human immunodeficiency virus type 1 primary isolates toantibodies and CD4-based reagents is independent of coreceptor usage.J Virol. 1998;72:1876-1885.
FREE FULL TEXT
79. Zwick MB, Labrijn AF, Wang M, et al. Broadly neutralizingantibodies targeted to the membraneproximal external region of humanimmunodeficiency virus type 1 glycoprotein gp41. JVirol. 2001; 75:10892-10905.
FREE FULL TEXT
80. Trkola A, Kuster H, Rusert P, et al. Delay of HIV-1 rebound aftercessation of antiretroviral therapy through passive transfer of humanneutralizing antibodies. Nat Med.2005;11:615-622.
PUBMED
81. Husson RN, Chung Y, Mordenti J, et al. Phase I study ofcontinuous-infusion soluble CD4 as a single agent and in combination with oraldideoxyinosine therapy in children with symptomatic human immunodeficiencyvirus infection. J Pediatr.1992;121:627-633.
PUBMED
82. Schacker T, Coombs RW, Collier AC, et al. The effects of high-doserecombinant soluble CD4 on human immunodeficiency virus type 1 viremia.J Infect Dis.1994;169:37-40.
FREE FULL TEXT
83. Jacobson JM, Israel RJ, Lowy I, et al. Treatment of advanced humanimmunodeficiency virus type 1 disease with the viral entry inhibitor PRO 542.Antimicrob Agents Chemother.2004;48:423-429.
FREE FULL TEXT
84. Wang HG, Williams RE, Lin PF. A novel class of HIV-1 inhibitorsthat targets the viral envelope and inhibits CD4 receptor binding.Curr Pharm Des. 2004;10: 1785-1793.
PUBMED
85. Towie N. Drug firms donate compounds for anti-HIV gel.Nature. 2005;438:6-7.
PUBMED
86. Veazey RS, Klasse PJ, Schader SM, et al. Protection of macaquesfrom vaginal SHIV challenge by vaginally delivered inhibitors of virus-cellfusion. Nature.2005;438:99-102.
PUBMED
87. Lederman MM, Offord RE, Hartley O. Microbicides and other topicalstrategies to prevent vaginal transmission of HIV. Nat RevImmunol. 2006;6:371-382.
88. Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D.Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4lymphocytes. Nat Rev Drug Discov.2004;3:215-225.
PUBMED
89. Moyle GJ, Wildfire A, Mandalia S, et al. Epidemiology andpredictive factors for chemokine receptor use in HIV-1 infection. JInfect Dis. 2005;191:866-872.
FREE FULL TEXT
90. Hartley O, Gaertner H, Wilken J, et al. Medicinal chemistry appliedto a synthetic protein: development of highly potent HIV entry inhibitors.Proc Natl Acad Sci U S A.2004;101:16460-16465.
FREE FULL TEXT
91. Kish-Catalone TM, Lu W, Gallo RC, Devico AL. Preclinical evaluationof synthetic-2 RANTES as a candidate vaginal microbicide to target CCR5.Antimicrob Agents Chemother.2006;50:1497-1509.
FREE FULL TEXT
92. Simmons G, Clapham PR, Picard L, et al. Potent inhibition of HIV-1infectivity in macrophages and lymphocytes by a novel CCR5 antagonist.Science. 1997;276: 276-279.
FREE FULL TEXT
93. Lederman MM, Veazey RS, Offord R, et al. Prevention of vaginal SHIVtransmission in rhesus macaques through inhibition of CCR5.Science. 2004;306: 485-487.
FREE FULL TEXT
94. Feng Z, Dubyak GR, Lederman MM, Weinberg A. Cutting edge: humanbeta defensin 3: a novel antagonist of the HIV-1 co-receptor CXCR4.J Immunol.2006;177:782-786.
FREE FULL TEXT
95. Quinones-Mateu ME, Lederman MM, Feng Z, et al. Human epithelial-defensins 2 and 3 inhibit HIV-1 replication. AIDS.2003;17:F39-F48.
PUBMED
96. Roschke V, Clark S, Branco L, et al. Characterization ofa panel of novel human monoclonal antibodies that specifically antagonize CCR5and block HIV-1 entry. Presented at: 44th Annual InterscienceConference on Antimicrobial Agents and Chemotherapy; October 30-November 2,2004; Washington, DC. Abstract 2871.97. Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5receptor-based mechanism of action of 873140, a potent allostericnoncompetitive HIV entry inhibitor. Mol Pharmacol.2005;67:1268-1282.
FREE FULL TEXT
98. Marozsan AJ, Kuhmann SE, Morgan T, et al. Generation and propertiesof a human immunodeficiency virus type 1 isolate resistant to the smallmolecule CCR5 inhibitor, SCH-417690 (SCH-D). Virology.2005;338:182-199.
PUBMED
99. Westby M, van der Ryst E. CCR5 antagonists: hosttargeted antiviralsfor the treatment of HIV infection. Antivir ChemChemother. 2005;16:339-354.
100. GlaxoSmithKline terminates patient enrollment for phase 3studies of investigational HIV entry inhibitor Aplaviroc (GW873140)[press release]. Research Triangle Park, NC: GlaxoSmithKline; October 25,2005.101. Greaves W, Landovitz R, Fatkenheuer G, et al. Late virologicbreakthrough in treatment-naïve patients on a regimen of combivir plusvicriviroc. Presented at: XIII Conference on Retroviruses andOpportunistic Infections; February 5-8, 2006; Denver, Colo.Abstract 161LB.102. Schering-Plough provides update on phase II study ofvicriviroc: study continues in HIV treatmentexperienced patients[press release]. Kenilworth, NJ: Schering-Plough Corp; March 3,2006.103. Dean M, Jacobson LP, McFarlane G, et al. Reduced risk of AIDSlymphoma in individuals heterozygous for the CCR5-32 mutation.Cancer Res. 1999;59: 3561-3564.
FREE FULL TEXT
104. Castagna A, Biswas P, Beretta A, Lazzarin A. The appealing story ofHIV entry inhibitors: from discovery of biological mechanisms to drugdevelopment. Drugs.2005;65:879-904.
PUBMED
105. French MA, Price P, Stone SF. Immune restoration disease afterantiretroviral therapy. AIDS. 2004;18: 1615-1627.
PUBMED
106. Shelburne SA III, Hamill RJ. The immune reconstitution inflammatorysyndrome. AIDS Rev.2003;5:67-79.
PUBMED
107. Brenchley JM, Schacker TW, Ruff LE, et al. CD4 T cell depletionduring all stages of HIV disease occurs predominantly in the gastrointestinaltract. J Exp Med.2004;200:749-759.
FREE FULL TEXT
108. Heise C, Vogel P, Miller CJ, Lackner A, Dandekar S. Distribution ofSIV infection in the gastrointestinal tract of rhesus macaques at early andterminal stages of AIDS. J Med Primatol.1993;22:187-193.
PUBMED
109. Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, RoedererM. Massive infection and loss of memory CD4T cells in multiple tissues duringacute SIV infection. Nature.2005;434:1093-1097.
PUBMED
110. Veazey RS, DeMaria M, Chalifoux LV, et al. Gastrointestinal tractas a major site of CD4T cell depletion and viral replication in SIV infection.Science. 1998;280: 427-431.
FREE FULL TEXT
111. Lange CG, Lederman MM, Medvik K, et al. Nadir CD4T-cell count andnumbers of CD28CD4 T-cells predict functional responses to immunizations inchronic HIV-1 infection. AIDS.2003;17:2015-2023.
PUBMED
112. Valdez H, Smith KY, Landay A, et al; ACTG 375 Team and AIDSClinical Trials Group. Response to immunization with recall and neoantigensafter prolonged administration of an HIV-1 protease inhibitorcontainingregimen. AIDS.2000;14:11-21.
PUBMED
113. Deeks SG. Challenges of developing R5 inhibitors in antiretroviralnaive HIV-infected patients. Lancet.2006;367:711-713.
PUBMED
ARTICLE EN RAPPORT
Biologie et rôle de CCR5 dans l'infection à VIH et dans son traitement
Michael M. Lederman, Adam Penn-Nicholson, Michael Cho, et Donald Mosier
JAMA. 2006;296:815-826.
Résumé
| Texte Complet
|