|
|
PAGES DU PRATICIEN
Hypertension pulmonaireUne complication de plus en plus reconnue des anémies hémolytiques héréditaires et de l'infection par le VIH
Christopher F. Barnett, MD, MPH;
Priscilla Y. Hsue, MD;
Roberto F. Machado, MD
RÉSUMÉ
| |
Les soins de santé modernes ont considérablement
augmenté la longévité des patients présentant une
anémie hémolytique congénitale (telles que la
drépanocytose et la thalassémie) et/ou une infection par le
virus de l'immunodéficience humaine (VIH). On estime que 10% des
patients présentant des hémoglobinopathies et 0.5% des patients
présentant une infection par le VIH développent une hypertension
pulmonaire modérée à sévère. L'hypertension
pulmonaire est une maladie implacablement progressive aboutissant à une
insuffisance cardiaque droite et à la mort. Dans le monde, il existe
environ 30 millions de patients atteints de drépanocytose ou de
thalassémie et 40 millions de patients atteints par le VIH. Vu la
prédominance de l'atteinte vasculaire pulmonaire dans ces populations,
la drépanocytose et l'infection par le VIH peuvent être les
causes les plus communes d'hypertension pulmonaire dans le monde. Dans cette
revue, les données disponibles sur l'épidémiologie,
l'hémodynamique, les mécanismes, et les stratégies
thérapeutiques dans ces maladies sont récapitulés.
Puisque la thérapie est susceptible de réduire la
morbidité et de prolonger la survie, des efforts pour dépister,
diagnostiquer, et soigner ces patients sont un objectif sanitaire
global.
JAMA.
2008;299(3):324-331
PRESENTATION DU CAS
L'évaluation complémentaire indiquait une faible
probabilité à la scintigraphie de ventilation/perfusion, des
tests de la fonction pulmonaires montrant une faible capacité de
diffusion de l'oxyde de carbone, et une positivité au test
sérique par immuno-absorption enzymatique et Western blot pour le virus
d'immunodéficience humaine (VIH), avec un nombre de CD4 T de 342/µl
(normale: 300-800/µl) avec une charge virale de 49 500 copies/ml (normale :
indétectable). Il marchait 240 m en 6 minutes (normale : 720 m). Un
échocardiogramme montrait une dilatation ventriculaire droite et un
dysfonctionnement systolique, une vitesse du flux de régurgitation
tricuspide (TRV) de 3.7 m/s (pression systolique artérielle pulmonaire
estimée de 60 mm Hg; normale <30 mm Hg), et une fonction
ventriculaire gauche normale (FIGURE
1 et FIGURE 2)
(voir la vidéo à
http://jama.com/content/full/299/3/324/DC1).
La cathétérisation du coeur droit montrait une pression
artérielle systolique pulmonaire de 64 mm Hg et une pression
artérielle diastolique de 39 mm Hg (moyenne, 47 mm Hg; normale <25
mm Hg), une pression capillaire pulmonaire de 14 mm Hg (normale <15 mm Hg),
et une saturation d'oxygène du sang veineux mixte de 58% (normale est
>75%). Son débit cardiaque était de 6 l/min (normale : 4-6
l/min) avec une résistance vasculaire pulmonaire de 440
dynes*s*cm-5 (normale :<240
dynes*s*cm-5). Les options thérapeutiques sont
envisagées.
|
|
|
|
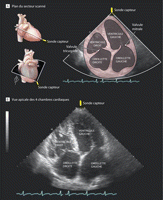
Figure 1.. Caractéristiques écho-cardiographiques d'hypertension
pulmonaire dans la drépanocytose (Cas du patient)
A, Plan du secteur scanné et contour des chambres cardiaques. B, La
vue apicale des 4 cavités démontre l'agrandissement de toutes
les chambres cardiaques caractéristique des patients chroniquement
anémiques atteints de drépanocytose et d'hypertension
pulmonaire. Cette vue démontre également la présence
d'une hypertrophie ventriculaire droite. (Voir vidéo à
http://jama.com/cgi/content/full/299/3/324/DC1.)
|
|
|
|
|
|
|
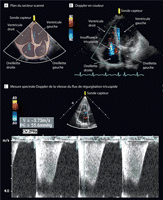
Figure 2.. Evaluation par Echo Doppler de la pression systolique de l'artère
pulmonaire (Cas du patient)
A, Plan du secteur scanné B, Doppler Couleur démontrant la
régurgitation tricuspide (bleue). C, Mesure spectrale Doppler de la
vitesse du flux de régurgitation tricuspide pour évaluer la
pression systolique de l'artère pulmonaire. La figure démontre
une vitesse du flux de régurgitation tricuspide maximale
d'approximativement 3.7 m/s. Un échantillon de la vitesse maximale du
flux de régurgitation tricuspide est employé pour estimer le
gradient de pression ventricule droit-pression systolique atriale (55.6 mm Hg
dans la figure) en utilisant l'équation modifiée de Bernoulli
(4x [vitesse du flux de régurgitation tricuspide]
2). La pression
systolique de l'artère pulmonaire est mesurée en ajoutant le
gradient de pression dérivé de Bernoulli à une estimation
de la pression atriale droite moyenne. (Voir la vidéo à
http://jama.com/cgi/content/full/299/3/324/DC1.)
|
|
|
L'hypertension pulmonaire est un processus pathologique lié à
une augmentation de la pression artérielle pulmonaire moyenne. Ce peut
être la conséquence de plusieurs états pathologiques dont
l'hypertension artérielle pulmonaire, les atteintes cardiaques gauches,
les affections pulmonaires liées à une hypoxie, et les embolies
pulmonaires chroniques.
Spécifiquement, l'hypertension artérielle pulmonaire se
définit par une augmentation de la pression artérielle
pulmonaire moyenne liée à l'atteinte artérielle de la
vascularisation pulmonaire. L'hypertension artérielle pulmonaire peut
être idiopathique, familiale, ou secondaire à une série de
pathologies telles que les maladies du tissu conjonctif, les shunts
congénitaux gauche-droit, certaines drogues et toxines, les
hémoglobinopathies, la cirrhose hépatique, ou l'hypertension
artérielle pulmonaire due à une infection par le
VIH.1 L'hypertension
artérielle pulmonaire mène à une augmentation progressive
de la pression artérielle pulmonaire moyenne et de la résistance
vasculaire pulmonaire, et à une diminution du débit cardiaque.
Lors de la progression de la maladie, la pression artérielle pulmonaire
peut se normaliser ou diminuer tandis que survient une insuffisance cardiaque
droite avec une chute du débit cardiaque, conduisant finalement
à une limitation de l'effort et une mort progressive.
L'étiologie de l'élévation des pressions pulmonaires
étant mixte (hypertension artérielle pulmonaire et
dysfonctionnement du coeur gauche) dans la SCD, elle est
désignée sous le nom d'hypertension pulmonaire dans le reste de
cet article.
Les patients atteints d'hypertension pulmonaire ont un tableau clinique
fait de dyspnée à l'effort, de fatigue, et de syncope. Beaucoup
de patients aux stades précoces de la maladie attribueront ces
symptômes à un déconditionnement et ne consultent pas
lorsque les symptômes sont observés pour la première fois.
Lorsqu'ils consultent, les symptômes sont souvent attribués
à une maladie chronique, retardant aussi le diagnostic d'hypertension
pulmonaire par des mois ou des années. L'hypertension pulmonaire est
une entité qu'il est médicalement important de
reconnaître, car elle compromet la qualité de vie et la survie,
et le pronostic pourrait être amélioré par une
intervention thérapeutique.
Les maladies hémolytiques et le VIH pourraient représenter
des causes importantes d'hypertension pulmonaire. Trente millions d'individus
dans le monde entier ont une
SCD2 et 40 millions
d'individus dans le monde entier ont une infection par le
VIH.3
Étant donné que l'hypertension pulmonaire se produit chez 10%
à 30% des patients ayant une
hémoglobinopathie4
et chez 0.5% des patients avec
VIH,5 il peut y
avoir 3 à 9 millions de patients avec hémoglobinopathie et 200
000 patients avec infection par le VIH affectés dans le monde par une
hypertension pulmonaire. Si l'histoire naturelle de l'hypertension pulmonaire
est aussi péjorative dans les SCD et l'infection par le HIV qu'elle est
dans d'autres populations de patients, l'hypertension pulmonaire deviendra un
problème majeur de santé au sein de ces populations.
HYPERTENSION PULMONAIRE ASSOCIEE A UNE ANEMIE HEMOLYTIQUE
Remarquablement, pratiquement chaque cause d'anémie
hémolytique, y compris la SCD, les thalassémies mineure et
majeure, la sphérocytose héréditaire et la stomatocytose,
l'hémoglobinurie nocturne paroxystique, l'anémie
hémolytique par hémoglobine de Mayence, l'anémie
hémolytique micro-angiopathique, le paludisme, l'hémolyse des
valves cardiaques artificielles, les dispositifs d'assistance ventriculaire
gauche, et les procédures de pontage cardio-pulmonaire, a
été associée à une hypertension pulmonaire
(récemment passée en revue par Gladwin et Kato,
6 liste
complète des références à
http://www.cc.nih.gov/ccmd/resources/bibliography.html).
En revanche, il n'y a aucune étude sur l'hypertension pulmonaire dans
les anémies des maladies chroniques ou ferriprives. Ces observations
soulignent le rôle critique de l'hémolyse dans le
développement de l'hypertension pulmonaire.
Epidémiologie et signification clinique
Les études rétrospectives de patients présentant une
SCD ou une thalassémie indiquent que la prédominance de
l'hypertension pulmonaire cliniquement significative varie de 10% à
50%.4,7
Les résultats histologiques montrant une hypertension pulmonaire sont
encore bien plus communs dans les SCD—75% des patients ayant des signes
à
l'autopsie.7 Ces
données sont corroborées par les études prospectives sur
le dépistage de l'hypertension pulmonaire des Instituts Nationaux de
Santé, qui ont constaté que 32% des patients ayant des SCD
avaient une élévation des pressions systoliques
artérielles pulmonaires (définies par une TRV  2.5 m/s, ce
qui correspond à une pression systolique artérielle pulmonaire
d'approximativement 30 mm Hg) et 9% avaient une élévation des
pressions modérée à sévère (définies
par une TRV de 3.0 m/s, ce qui correspondent à une pression
systolique artérielle pulmonaire d'approximativement 41 mm
Hg).7 Des taux
semblables ont été trouvés lors d'études
écho-cardiographiques de patients atteints par une
β-thalassémie ou une
SCD.8,9
La mesure rétrospective du N-terminal pro-brain natriuretic peptide,
une pro-hormone libérée à l'effort par les myocardes
ventriculaires droit et gauche soumis à un stress d'effort, prouve que
30% des patients atteint de SCD avaient une élévation des taux,
suggérant la présence possible d'une hypertension
pulmonaire.10 2.5 m/s, ce
qui correspond à une pression systolique artérielle pulmonaire
d'approximativement 30 mm Hg) et 9% avaient une élévation des
pressions modérée à sévère (définies
par une TRV de 3.0 m/s, ce qui correspondent à une pression
systolique artérielle pulmonaire d'approximativement 41 mm
Hg).7 Des taux
semblables ont été trouvés lors d'études
écho-cardiographiques de patients atteints par une
β-thalassémie ou une
SCD.8,9
La mesure rétrospective du N-terminal pro-brain natriuretic peptide,
une pro-hormone libérée à l'effort par les myocardes
ventriculaires droit et gauche soumis à un stress d'effort, prouve que
30% des patients atteint de SCD avaient une élévation des taux,
suggérant la présence possible d'une hypertension
pulmonaire.10
L'hypertension pulmonaire augmente le risque de décès chez
les patients atteints de SCD. Dans l'étude de l'Institut National de
Santé, comparés aux patients ayant une TRV inférieure
à 2.5 m/s (pression systolique artérielle pulmonaire de 30 mm
Hg), les taux relatifs de décès avec une TRV de 2.5 à 2.9
m/s (pression systolique artérielle pulmonaire de 30-37 mm Hg) et
supérieure à 3.0 m/s (pression systolique artérielle
pulmonaire de 41 mm Hg) étaient respectivement de 4.4 (intervalle de
confiance à 95% [IC], 1.6-12.2 ; P<0.001) et 10.6 (IC 95%, 3.3-33.6
; P<0.001), pour une médiane de suivi de 27
mois.7,11
À l'appui de ces résultats, De Castro et
al9 ont
constaté que 6 patients sur 42 (14%) présentant une hypertension
pulmonaire et 2 patients sur 83 (2%) sans hypertension pulmonaire sont
décédés au cours d'une période de deux ans de
suivi. De même, dans l'étude d'Ataga et collaborateurs, 89
patients sur 36 présentant une hypertension pulmonaire et 1 patient sur
57 sans hypertension pulmonaire sont décédés au cours
d'une période de suivi de 2.5 ans (risque relatif, 9.24 ; IC 95%,
1.20-73.30). A ce jour, il n'y a aucune étude ayant
évalué l'impact de l'hypertension pulmonaire sur la survie des
patients présentant une thalassémie ou une autre
hémoglobinopathie.
Pathogenèse
Chez les patients atteints de SCD, la sévérité de
l'hémolyse est corrélée à la
sévérité de l'hypertension
pulmonaire.7 Ceci
suggère que l'hémolyse est liée mécaniquement
à l'hypertension pulmonaire. Une telle relation est biologiquement
plausible, car l'hémoglobine libre inactive l'oxyde nitrique
intrinsèque vaso-dilatateur. L'hémolyse libère
également l'arginase, qui dégrade la L-arginine, substrat pour
la synthèse d'oxyde nitrique,
12 entraînant
un état de diminution de la biodisponibilité et de <<
résistance >> à la vasodilatation dépendante de l'oxyde
nitrique.13
L'hémolyse et la diminution de la biodisponibilité de l'oxyde
nitrique induisent également une activation des plaquettes, une
génération de thrombine
14, et du facteur
activation
tissulaire.15 Ces
facteurs contribuent tous à un plus grand risque de thrombose.
L'accumulation de l'hème et du fer redox actifs des globules rouges
lysés contribue par ailleurs à la génération des
espèces réactives de l'oxygène (reactive oxygen species)
qui peuvent ainsi aggraver les lésions dues au syndrome
d'ischémie-reperfusion, à la thrombose, et les réponses
prolifératives endothéliales et des muscles
lisses.16 Ainsi,
l'hémolyse déclenche des processus multiples qui pourraient
entraîner une hypertension pulmonaire.
D'autres facteurs pourraient contribuer à l'hypertension pulmonaire
dans la SCD. Chez les patients avec SCD, à la fois lors des phases
stables et des crises douloureuses vaso-occlusives, les taux plasmatiques
d'endothéline-1 sont
augmentés.17
In vitro, les érythrocytes dans la drépanocytose augmentent la
production d'endothéline-1 par les cellules endothéliales
humaines en culture. En outre, l'antagonisme du récepteur A de
l'endothéline supprime dans la drépanocytose les effets
vasoconstricteurs de la média des cellules endothéliales
pulmonaires exposées aux érythrocytes sur des anneaux
aortiques.17 Ces
observations suggèrent toutes une augmentation de l'activité
vaso-constrictrice dans la SCD et les autres troubles hémolytiques.
La perte de la fonction splénique peut déclencher une
activation des plaquettes, favorisant l'adhésion de micro-thromboses
pulmonaires et des globules rouges sur l'endothélium.18 La rate joue
également une fonction critique dans l'élimination des
érythrocytes sénescents et endommagés. Après
splénectomie, le taux d'hémolyse intra-vasculaire
augmente.19,20
Le dysfonctionnement splénique peut ainsi produire une série de
processus qui contribuent à l'hypertension pulmonaire.
Historiquement, on a pensé que l'hypertension pulmonaire chez les
patients avec SCD dérivait des épisodes
réitérés de crises vaso-occlusives pulmonaires et du
syndrome thoracique aigu entraînant une fibrose pulmonaire, une
obstruction vasculaire, et une hypoxie chronique.
Cependant, cette association n'a pas été appuyée par
les études épidémiologiques car le nombre
d'épisodes de crises vaso-occlusives et de syndromes aigus thoraciques
n'est pas associé à l'hypertension pulmonaire,
7,10
En outre, l'hypertension pulmonaire se produit dans d'autres désordres
hémolytiques tels que la thalassémie, qui n'est pas
associée à une vaso-occlusion ou à un syndrome
thoracique.4,13
La théorie que les désordres vaso-occlusifs ou le syndrome
thoracique contribuent ainsi à une hypertension pulmonaire peut ne pas
être valide.
Les patients présentant des désordres hémolytiques
chroniques deviennent symptomatiques lorsque les pressions artérielles
pulmonaires moyennes atteignent 30 à 40 mm
Hg.21 En revanche,
les patients présentant d'autres formes d'hypertension pulmonaire ne
sont pas symptomatiques jusqu'à ce que la pression artérielle
pulmonaire moyenne atteigne 50 à 60 mm
Hg.22 Les patients
présentant des désordres hémolytiques chroniques ont
également des augmentations légères de la
résistance vasculaire pulmonaire et une élévation
légère co-existante de la pression capillaire pulmonaire,
suggérant une insuffisance cardiaque gauche. Les données de
cathétérisation du coeur droit prouvent que 54% de patients avec
SCD ont une hypertension artérielle pulmonaire tandis qu'avec une
atteinte cardiaque gauche, une hypertension veineuse pulmonaire secondaire se
voit chez 46%.21
Sachdev et al23 ont
constaté que 47% des patients ont une hypertension pulmonaire, un
dysfonctionnement diastolique, ou les deux (29% avaient seulement une
hypertension pulmonaire, 11% un dysfonctionnement diastolique et hypertension
pulmonaire, et 7% un dysfonctionnement diastolique seul). L'hypertension
pulmonaire et le dysfonctionnement diastolique ont été
associés à un risque relatif de décès de 5.1 (IC
95%, 2.0-13.3) et 4.8 (IC 95%, 1.9-12.1), respectivement, alors que le risque
relatif de décès en présence des deux était de
12.0 (IC 95%,
3.8-38.1).23 Ces
données suggèrent que l'hypertension pulmonaire et le
dysfonctionnement diastolique surgissent indépendamment et comportent
un risque additif de mortalité.
Manifestations cliniques et évaluation
Les patients présentant une anémie maintiennent un
débit cardiaque de repos compensatoire élevé pour assurer
une livraison adéquate d'oxygène. En soi, ils semblent
être même médiocrement tolérants à de faibles
augmentations additionnelles de la résistance vasculaire pulmonaire qui
peut être associée à l'effort ou aux crises
hémolytiques.24
Comparé au SCD seul, les patients avec SCD et hypertension pulmonaire
ont une diminution au test de marche de 6 minutes (moyenne [ES], 320[20] mvs
435[31]m ; P=0.002) et pic de consommation de l'oxygène au test
d'effort cardio-pulmonaire (moyen [ES], 41% [2%] de la consommation
théorique contre 50% [3%] ;
P=0.02).21 Les
patients présentant une hypertension artérielle pulmonaire
provenant d'autres étiologies et une pression artérielle
pulmonaire moyenne plus élevée (54 mm Hg) ont également
mieux exécuté le test de marche sur 6 minutes (398 m) le test
d'effort (46% de la consommation
théorique).25
Chez les patients présentant des désordres
hémolytiques chroniques, l'intolérance à l'effort est
souvent attribuée à une anémie ou à une affection
pulmonaire plutôt qu'à l'hypertension pulmonaire.
L'évaluation diagnostique de l'intolérance à l'effort
devrait inclure une recherche intensive des autres pathologies qui peuvent
contribuer à une hypertension pulmonaire, telle que la surcharge de
fer, les affections hépatiques chroniques, le VIH, l'hypoxémie
nocturne, et la maladie thrombo-embolique.
Une cathétérisation diagnostique du coeur droit est
essentielle.26 Le
test de marche de 6 minutes est inversement corrélé à la
sévérité de l'hypertension
pulmonaire,21 et la
thérapie spécifique de l'hypertension pulmonaire améliore
la distance de
marche.27 Aussi, le
test de marche de 6 minutes est un substitut utile de la capacité
fonctionnelle dans cette population de patients.
HYPERTENSION ARTERIELLE PULMONAIRE ASSOCIEE AU VIH
Epidémiologie et signification clinique
La prévalence de l'hypertension artérielle pulmonaire
associée au VIH a été estimée être entre
0.06% et 2%.5
L'évaluation la plus complète de l'hypertension
artérielle pulmonaire associée au VIH a été
réalisée dans une étude de cohorte prospective de 10 547
patients avec VIH et a montré une prévalence de l'hypertension
artérielle pulmonaire de 0.21% (IC 95%,
0.12%-0.30%).28
Dans cette étude, l'hypertension artérielle pulmonaire
était diagnostiquée par cathétérisation du coeur
droit. Deux études ont évalué l'impact de la
thérapie antirétrovirale sur la prévalence de
l'hypertension artérielle pulmonaire, mais sont arrivées
à des conclusions opposées sur le fait de savoir si la
thérapie antirétrovirale est associée à une
réduction de l'hypertension artérielle
pulmonaire.5
Preuve de la présence d'une maladie précoce
Une étude écho-cardiographique rétrospective chez les
patients de la clinique VIH des National Institutes of Health a prouvé
que 9.3% de patients avaient une TRV de 2.5 m/s ou plus (pression systolique
artérielle pulmonaire de 30 mm Hg) et 0.4% une TRV de 3.0 m/s ou plus
(pression systolique artérielle pulmonaire de 41 mm
Hg).29 En prenant
en considération la TRV maximale pour chaque patient au cours de la
période d'observation, 16.4% avaient une TRV de 2.5 m/s ou plus et 0.7%
avaient une TRV de 3.0 m/s ou plus au moins à une occasion. L'analyse
univariée montrait des associations significatives entre une TRV
élevée et l'âge (P=0.05), le sexe masculin (P=0.03), un
nombre inférieur de CD4 T (r=-0.14 ; P=0.02), et l'absence d'exposition
à l'interleukine 2 (P=0.02). La prévalence d'une TRV
élevée de 0.4% à 0.7% parmi la population de
l'étude est compatible à ce qui a été
précédemment rapporté,
5 de même que
les associations à un âge plus avancé et au sexe
masculin.
Dans une analyse semblable de patients du General Hospital de San
Francisco, la TRV et la pression atriale droite ont été
utilisées comme moyen d'évaluation de la pression systolique
artérielle pulmonaire chez 186 individus atteints d'une infection par
le VIH et 36 témoins d'âge comparable sans infection par le
VIH.30 Les
personnes ayant une infection par le VIH avaient une pression systolique
artérielle pulmonaire plus élevée que les témoins
(médiane [extrêmes interquartiles], 26[19-31] mm Hg par rapport
à 21 [17-26] mm Hg; P=0.001). Une pression systolique artérielle
pulmonaire supérieure à 30 mm Hg et 40 mm Hg a été
trouvée chez 30% des patients avec VIH comparés à 6% des
témoins (P=0.003), et chez 6% des patients avec VIH comparés
à 0% des témoins (P=0.28), respectivement. Après
ajustement sur l'âge, le sexe, le tabagisme, et l'utilisation
intraveineuse de drogues, les individus avec VIH avaient 5.5 fois plus de
chances d'avoir une pression systolique artérielle pulmonaire
supérieure à 30 mm Hg (P=0.02).
La prévalence d'une pression systolique artérielle pulmonaire
élevée dans la cohorte du General Hospital de San Francisco (6%)
est sensiblement plus importante que celle dans la cohorte des National
Institutes of Health (0.4%-0.7%), mais est semblable à celle
trouvée dans une étude allemande de dépistage
(7.5%).31 La raison
de cette différence pourrait être due à la
démographie, au mode de transmission, ou à l'utilisation
intraveineuse de drogue. Bien que les preuves liant l'abus de drogues au
développement de l'hypertension pulmonaire dans l'infection par le VIH
soient peu concluantes,
32,33
la prévalence de l'utilisation intraveineuse de drogues dans la cohorte
du General Hospital de San Francisco était de 38% par rapport à
4% dans celle des National Institutes of Health.
L'hypertension artérielle pulmonaire augmente la mortalité
des patients atteints d'une infection par le VIH comme elle le fait dans
d'autres populations de patients. Dans une étude cas-témoins, le
risque relatif de décès chez les individus ayant une infection
par le VIH et hypertension pulmonaire étaient de 2.1 (IC 95%, 1.0-4.5 ;
P<0.05) comparés aux individus avec VIH mais sans hypertension
pulmonaire.34 En
outre, la survie médiane des patients présentant une
hypertension pulmonaire était de 1.3 an comparée à 2.6
ans dans le groupe sans hypertension pulmonaire (P<0.05). Bien que ces
données aient 10 années, elles représentent
peut-être mieux l'histoire naturelle des effets de l'hypertension
pulmonaire sur la mortalité chez les patients ayant une infection par
le VIH parce que la population est relativement uniforme. Seule une faible
proportion de patients a été soignée avec un traitement
antirétroviral (zidovudine seulement) et aucun patient n'a reçu
de traitement de l'hypertension pulmonaire. Aussi, bien que la
mortalité associée au VIH soit sensiblement
améliorée par la thérapie antirétrovirale
hautement active, les données de mortalité de l'ère avant
la thérapie antirétrovirale hautement active et avant la
thérapie spécifique de l'hypertension pulmonaire peuvent fournir
une description plus claire des effets de l'hypertension artérielle
pulmonaire associée au VIH sur la survie.
Pathogenèse
Les patients atteints par le VIH avec hypertension artérielle
pulmonaire ont des lésions plexogéniques semblables aux patients
présentant d'autres maladies associées à une hypertension
artérielle
pulmonaire.35 Il
n'y a eu aucune association constante entre l'hypertension artérielle
pulmonaire et le nombre de CD4 T (contrairement aux résultats de la
cohorte des National Institutes of Health) ou la charge virale
VIH.36
Il n'a jamais été démontré que le virus de
l'immunodéficience humaine infectait directement les cellules
endothéliales vasculaires
pulmonaires,37,38
mais des antigènes viraux VIH sont présents dans
l'endothélium pulmonaire et peuvent directement stimuler une apoptose,
une croissance, et une prolifération
anormales.39 Il a
été montré que la glycoprotéine 120, une
protéine virale nécessaire pour la liaison et l'entrée du
VIH dans les macrophages, cible les cellules endothéliales du poumon
humain, augmente les marqueurs de l'apoptose, et stimule la
sécrétion
d'endothéline-1.37
L'antigène du facteur Nef, critique pour l'entretien de la charge
virale et pour les interactions du signal dans la cellule hôte, a
été localisé dans de multiple types de cellules
pulmonaires et
vasculaires.40 In
vitro, les cellules endothéliales humaines exposées à Nef
démontrent un accroissement de l'apoptose, suivi par une
prolifération.41
Des primates infectés par le virus d'immunodéficience simiesque
exprimant la protéine Nef VIH développent des lésions
ressemblant aux lésions plexiformes, et l'antigène Nef est
discernable dans les tissus alvéolaires, vasculaires,
péri-vasculaires, et
lymphoïdes.40
Ainsi, le VIH pourrait plausiblement être lié à la
genèse de l'hypertension artérielle pulmonaire.
Il y a d'autres mécanismes par lesquels le VIH pourrait causer
l'hypertension artérielle pulmonaire. L'infection par le virus de
l'immunodéficience humaine induit un état inflammatoire
chronique et une activation et une dysrégulation immunitaire
persistante42 qui
pourraient indirectement induire la libération de cytokines et de
facteurs de croissance pro-inflammatoires pouvant produire une hypertension
artérielle
pulmonaire.43 Une
augmentation de l'expression du facteur de croissance dérivé des
plaquettes (PDGF), un stimulus efficace des cellules du muscle lisse et de la
croissance et migration des fibroblastes, a également été
notée dans le tissu pulmonaire des patients avec hypertension
artérielle pulmonaire associée au VIH.38 De même, le
facteur de croissance endothélial vasculaire A (VEGF A) induit une
perméabilité vasculaire et une prolifération des cellules
endothéliales et est produit par les cellules T infectées par la
VIH in vivo.44
Les mutations morphogénétiques osseuses du récepteur 2
des protéines (BMPR-2), liées à l'hypertension
artérielle pulmonaire familiale, ont comme conséquence une
diminution de la signalisation par
BMPR-2.45 La
protéine tat du VIH-1 (transactivateur transcriptionnel) réprime
l'expression du gène BMPR-2 dans les macrophages humains in vitro, en
interférant avec la régulation transcriptionnelle de BMP et de
BMPR-2.45 Il a
été également montré que la protéine
exogène tat active les cellules endothéliales, ayant pour
résultat une libération des facteurs de croissance,
45 soutenant
l'hypothèse que les protéines virales VIH pourraient induire une
fonction endothéliale anormale, menant à une hypertension
artérielle pulmonaire.
On a rapporté que l'herpès virus humain 8 est associé
histologiquement à une hypertension artérielle pulmonaire.
46 Cette
association n'a pas été uniformément confirmée par
d'autres.47
TRAITEMENT DE l'HYPERTENSION PULMONAIRE ASSOCIEE A UNE ANEMIE HEMOLYTIQUE ET UNE INFECTION PAR LE VIH
Le traitement de l'hypertension artérielle pulmonaire inclut
classiquement des diurétiques, des glycosides cardiaques, une
oxygénothérapie, une anti-coagulation par warfarine, et des
inhibiteurs calciques à fortes doses chez des patients
sélectionnés. Les prostanoïdes, les antagonistes de
l'endothéline, et les inhibiteurs de la phosphodiastérase-5 ont
été employés chez des patients symptomatiques pour
améliorer la tolérance à l'effort et
l'hémodynamique.48
Hypertension pulmonaire associée à une anémie hémolytique
Puisque l'hémolyse intra-vasculaire chronique est un
mécanisme central dans le développement de l'hypertension
pulmonaire, la thérapie de l'hémoglobinopathie sous-jacente doit
être optimisée. Pour la SCD, la thérapie par
hydroxyurée et les transfusions doivent être
optimisée.49,50
Une stratégie semblable semble raisonnable pour les autres types
d'anémies hémolytiques. Une étude récente
51 a prouvé
que l'hypertension pulmonaire avait été complètement
prévenue chez des patients ayant une thalassémie majeure
correctement transfusés et bien traités par chélateurs de
fer.
Des thérapies qui augmentent la biodisponibilité de l'oxyde
nitrique sont actuellement évaluées dans l'hypertension
artérielle pulmonaire. L'oxyde nitrique inhalé chronique dilate
sélectivement la vascularisation pulmonaire et inactive par oxydation
l'hémoglobine plasmatique
circulante.52
L'oxyde nitrique inhalé chronique est en cours d'investigation,
coûteux, et exige un système compliqué
d'administration.53
Il a été montré que la L-arginine diminue la pression
systolique artérielle pulmonaire de 15% dans une petite étude de
patients avec SCD54
et semble prometteuse comme autre approche destinée à
améliorer la biodisponibilité de l'oxyde nitrique. Le
sildénafil agit en empêchant le métabolisme de la
guanosine monophosphate cyclique, le deuxième messager qui
régule les effets de l'oxyde nitrique. Dans une série de cas
récemment
publiée,55 7
patients présentant diverses formes de thalassémie avec
hypertension pulmonaire grave ont été traités par
sildénafil durant 4 semaines à 48 mois avec une
amélioration de la TRV, de la classe fonctionnelle de la New York Heart
Association, et du test de marche de 6 minutes. Douze patients de la cohorte
des National Institutes of Health ont été soignés par
sildénafil pendant une moyenne de 6
mois.27 La pression
systolique artérielle pulmonaire moyenne avait diminué de 9 mm
Hg (IC 95%, 0.3-17.0 mm Hg; P=0.05), le test de marche de 6 minutes s'est
amélioré de 78 m (IC 95%, 40-117 m ; P=.003), le N-terminal
pro-brain natriuretic peptide a diminué de 448 pg/ml
(P=0.002).27 Ces
résultats sont d'une manière encourageante et suggèrent
le besoin d'un essai à plus grande échelle soutenue par le
National Heart, Lung, and Blood Institute.
Hypertension artérielle pulmonaire associée au VIH
Il n'y a eu aucune grande étude sur les effets de la thérapie
antirétrovirale sur l'hypertension artérielle pulmonaire
associée au VIH. L'époprosténol et le bosentan semblent
être efficaces en termes d'amélioration des symptômes et de
l'hémodynamique, mais les études jusqu'ici ont concerné
peu de patients.56
Le sildénafil a été également employé avec
des résultats
encourageants.56
Cependant, les interactions entre le sildénafil et les agents
antirétroviraux, particulièrement le ritonavir, peuvent
augmenter dangereusement le sildénafil et une hypotension symptomatique
peut se produire si des ajustements de dose ne sont pas faits.
Ainsi, l'hypertension artérielle pulmonaire associée au VIH a
été traitée efficacement chez quelques patients, mais les
effets de ces agents sur l'histoire naturelle de l'hypertension
artérielle pulmonaire restent à déterminer dans cette
population de patients.
CONCLUSIONS
Dans le cas présenté, la SCD du patient a été
contrôlée par des exsanguino-transfusions et une association de
traitements antirétroviraux (stavudine, lamivudine, et
éfavirenz). L'hémoglobine du patient est restée stable,
son nombre de CD4 T a grimpé jusqu'à 400/µl, et sa charge
virale a diminué à 400 copies/ml. Son hypertension pulmonaire a
été traitée par sildénafil. Il a
éprouvé une amélioration symptomatique et le test de
marche de 6 minutes a grimpé jusqu'à 430 M. Un
échocardiogramme de suivi a montré une élévation
persistante de la TRV de 3.2 m/s (pression systolique artérielle
pulmonaire : 46 mm Hg), mais la taille du ventricule droit et la fonction
systolique se sont améliorées. Son état clinique est
demeuré stable pendant approximativement 1,5 année, puis il a
commencé à développer une aggravation de la
dyspnée d'effort liée à la progression de l'hypertension
pulmonaire, exigeant l'addition de bosentan à son protocole. Il a
éprouvé une amélioration initiale des symptômes, de
la capacité fonctionnelle, et des pressions pulmonaires. Cependant,
après 2 ans il a développé une insuffisance cardiaque
droite progressive. Un traitement traitement par prostacycline intraveineuse
ou sous-cutanée a été proposé, mais a
été refusé par le patient. De l'iloprost inhalé a
été initié, mais peu de temps après le patient est
décédé soudainement chez lui. L'identification de
l'hypertension pulmonaire de ce patient et une thérapie
appropriée a ainsi permis une amélioration symptomatique.
L'amélioration de la survie peut être déterminée
par des études à plus grande échelle.
Comme les patients ayant des troubles hémolytiques et une infection
par le VIH survivent plus longtemps, de nouveaux processus pathologiques qui
menacent leur qualité de vie et la survie sont susceptibles
d'émerger. L'hypertension pulmonaire semble correspondre à ce
genre de menace. Le dépistage de l'hypertension pulmonaire dans ces
populations est justifié. Puisque des traitements, pouvant changer la
progression de l'hypertension pulmonaire, deviennent disponibles, les
investigations doivent être élargies pour comprendre comment
l'hypertension pulmonaire et sa thérapie peuvent être
contrôlées.
Informations sur les auteurs
Correspondance: Roberto F. Machado, MD, Pulmonary and Vascular Medicine
Branch, National Heart, Lung, and Blood Institute, National Institutes of
Health, Clinical Center, Bldg 10-CRC, Room 5-5140, Bethesda, MD 20892
(robertom{at}nhlbi.nih.gov).
Rédacteur en chef de la section Tables Rondes: David S.
Cooper, MD, Rédacteur associé, JAMA.
Contributions des auteurs : Le Dr Machado a eu un accès
complet à toutes les données de l'étude et accepte la
responsabilité de l'intégrité des données et de
l'exactitude de l'analyse des données.
Conception et schéma de l'étude: Barnett,
Machado.
Recueil des données: Barnett, Hsue, Machado.
Analyse et interprétation des données: Barnett,
Hsue, Machado.
Recueil du manuscrit: Barnett, Hsue, Machado.
Revue critique du manuscrit: Barnett, Hsue, Machado.
Obtention du financement: Hsue.
Aide administrative, technique et matérielle: Barnett,
Hsue, Machado.
Liens financiers: Le Dr Hsue a déclaré avoir
reçu une bourse d'Actelion. Aucun autre auteur n'a rapporté de
lien financier.
Financement/Soutien: Le Dr. Hsue est soutenu par un prix de
développement scientifique et clinique de la Doris Duke Charitable
Fondation et par une subvention Grant-in-Aid de l'association
américaine du coeur.
Role du sponsor: Les sponsors n'ont joué aucun rôle
dans la conception et la conduite de l'étude ; la collection, la
gestion, l'analyse, et l'interprétation des données ; ni dans la
préparation, la revue, ou l'approbation du manuscrit.
Autres informations: Voir la vidéo en ligne à
http://www.jama.com/content/full/299/3/324/DC1.
Autres Contributions: Nous remercions Henry Masur, MD, et Mark Gladwin, MD,
pour leur examen critique du manuscrit et Mary K. Hall pour l'aide
éditoriale dans la préparation du manuscrit. Les Drs Masur et
Gladwin et Mme Hall travaillent dans le département de médecine
de soins critiques, centre clinique, National Institutes of Health, Bethesda,
Maryland. Le Dr. Gladwin et Mme Hall travaillent dans le secteur de
médecine pulmonaire et vasculaire, National Heart, Lung, and Blood
Institute, National Institutes of Health, Bethesda, Maryland. Aucune de ces
personnes n'a été rétribuée pour sa
contribution.
Affiliations des auteurs: Critical Care Medicine Department,
Clinical Center, and Pulmonary and Vascular Medicine Branch, National Heart,
Lung, and Blood Institute, National Institutes of Health, Bethesda, Maryland;
and Division of Cardiology, University of California, San Francisco.
BIBLIOGRAPHIE
| |
1. Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of
pulmonary hypertension. J Am Coll Cardiol.2004; 43(12)(suppl S):5S
-12S.
FREE FULL TEXT
2. Cavalli-Sforza LL, Menozzi P, Piazza A. The History and
Geography of Human Genes. Princeton, NJ: Princeton University
Press; 1994.3. World Health Organization; United Nations Joint Programme on
HIV/AIDS. AIDS Epidemic Update. Geneva, Switzerland:
World Health Organization; 2006.4. Machado RF, Gladwin MT. Chronic sickle cell lung disease: new
insights into the diagnosis, pathogenesis and treatment of pulmonary
hypertension. Br J Haematol.2005; 129(4):449
-464.
PUBMED
5. Zuber JP, Calmy A, Evison JM, et al. Pulmonary arterial
hypertension related to HIV infection: improved hemodynamics and survival
associated with antiretroviral therapy. Clin Infect
Dis.2004; 38(8):1178
-1185.
FREE FULL TEXT
6. Gladwin MT, Kato GJ. Cardiopulmonary complications of sickle cell
disease: role of nitric oxide and hemolytic anemia. Hematology (Am
Soc Hematol Educ Program). 2005;(1):51
-57.7. Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a
risk factor for death in patients with sickle cell disease. N Engl
J Med. 2004;350
(9):886
-895.
PUBMED
8. Ataga KI, Moore CG, Jones S, et al. Pulmonary hypertension in
patients with sickle cell disease: a longitudinal study. Br J
Haematol.2006; 134(1):109
-115.
PUBMED
9. De Castro LM, Jonassiant JC, Graham FL, Ashley-Koch A, Tellen MJ.
Pulmonary hypertension in SS, SC and S β thalassemia: prevalence,
associated clinical sydromes, and mortality. Blood.2004; 104(11):462A
.
10. Machado RF, Anthi A, Steinberg MH, et al. N-Terminal
pro-brain natriuretic peptide levels and risk of death in sickle cell disease.
JAMA.2006; 296(3):310
-318.
FREE FULL TEXT
11. Machado RF, Castro O. Sickle cell disease-associated pulmonary
hypertension: overview of clinical manifestations and emerging therapeutic
options. Adv Pulmonary Hypertens.2007; 6(1):16
-22.
12. Morris CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine
metabolism, hemolysis-associated pulmonary hypertension, and mortality in
sickle cell disease. JAMA.2005; 294(1):81
-90.
FREE FULL TEXT
13. Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of
intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism
of human disease. JAMA.2005; 293(13):1653
-1662.
FREE FULL TEXT
14. Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ.
Platelet activation in patients with sickle disease, hemolysis-associated
pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin.
Blood.2007; 110(6):2166
-2172.
FREE FULL TEXT
15. Hagger D, Wolff S, Owen J, Samson D. Changes in coagulation and
fibrinolysis in patients with sickle cell disease compared with healthy black
controls. Blood Coagul Fibrinolysis.1995; 6(2):93
-99.
PUBMED
16. Nath KA, Katusic ZS, Gladwin MT. The perfusion paradox and vascular
instability in sickle cell disease. Microcirculation.2004; 11(2):179
-193.
PUBMED
17. Ergul S, Brunson CY, Hutchinson J, et al. Vasoactive factors in
sickle cell disease: in vitro evidence for endothelin-1-mediated
vasoconstriction. Am J Hematol.2004; 76(3):245
-251.
PUBMED
18. Atichartakarn V, Likittanasombat K, Chuncharunee S, et al.
Pulmonary arterial hypertension in previously splenectomized patients with
β-thalassemic disorders. Int J Hematol.2003; 78(2):139
-145.
PUBMED
19. Atichartakarn V, Angchaisuksiri P, Aryurachai K, et al.
Relationship between hypercoagulable state and erythrocyte phosphatidylserine
exposure in splenectomized haemoglobin E/β-thalassaemic patients.
Br J Haematol.2002; 118(3):893
-898.
PUBMED
20. Kisanuki A, Kietthubthew S, Asada Y, Marutsuka K, Funahara Y,
Sumiyoshi A. Intravenous injection of sonicated blood induces pulmonary
micro-thromboembolism in rabbits with ligation of the splenic artery.
Thromb Res.1997; 85(2):95
-103.
PUBMED
21. Anthi A, Machado RF, Jison ML, et al. Hemodynamic and functional
assessment of sickle cell disease patients with pulmonary hypertension.
Am J Respir Crit Care Med.2007; 175(12):1272
-1279.
FREE FULL TEXT
22. Barst RJ, Rubin LJ, McGoon MD, Caldwell EJ, Long WA, Levy PS.
Survival in primary pulmonary hypertension with long-term continuous
intravenous prostacyclin. Ann Intern Med.1994; 121(6):409
-415.
FREE FULL TEXT
23. Sachdev V, Machado RF, Shizukuda Y, et al. Diastolic dysfunction is
an independent risk factor for death in patients with sickle cell disease.
J Am Coll Cardiol.2007; 49(4):472
-479.
FREE FULL TEXT
24. Machado RF, Kyle Mack A, Martyr S, et al. Severity of pulmonary
hypertension during vasoocclusive pain crisis and exercise in patients with
sickle cell disease. Br J Haematol.2007; 136(2):319
-325.
PUBMED
25. Barst RJ, Langleben D, Frost A, et al. Sitaxsentan therapy for
pulmonary arterial hypertension. Am J Respir Crit Care
Med.2004; 169(4):441
-447.
FREE FULL TEXT
26. McGoon M, Gutterman D, Steen V, et al. Screening, early detection,
and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical
practice guidelines. Chest.2004; 126(1)(suppl):14S
-34S.
PUBMED
27. Machado RF, Martyr S, Kato GJ, et al. Sildenafil therapy in
patients with sickle cell disease and pulmonary hypertension. Br J
Haematol.2005; 130(3):445
-453.
PUBMED
28. Sitbon OL, Sereni D, Raffi R, et al. Prevalence of pulmonary
arterial hypertension in HIV-positive out-patients in the highly active
antiretroviral therapy era. Proc Am Thorac Soc.2006; 3:A57
.
29. Barnett CF, Azad NS, Bishop MR, Barrett AJ, Gladwin MT, Machado RF.
Pulmonary arterial hypertension in HIV infection and stem cell transplant:
screening identified high prevalence of "pre-disease."
Proc Am Thorac Soc.2006; 3:A61
.
30. Hsue P, Waters D, Farah H, et al. HIV is associated with pulmonary
hypertension independent of known risk factors.
Circulation.2005; 112(suppl ll):97
.
31. Rosenkranz S, Steffen H, Vogel D, et al. HIV-associated pulmonary
hypertension in patients on HAART. Conf Retrovir Opportunistic
Infect. 2005; 12:874
.
32. Pellicelli AM, D'Ambrosio C, Vizza CD, et al. HIV-related pulmonary
hypertension: from pathogenesis to clinical aspects. Acta
Cardiol.2004; 59(3):323
-330.
PUBMED
33. Chin KM, Channick RN, Rubin LJ. Is methamphetamine use associated
with idiopathic pulmonary arterial hypertension?
Chest.2006; 130(6):1657
-1663.
PUBMED
34. Opravil M, Pechere M, Speich R, et al. HIV-associated primary
pulmonary hypertension: a case control study: Swiss HIV Cohort Study.
Am J Respir Crit Care Med.1997; 155(3):990
-995.
FREE FULL TEXT
35. Mehta NJ, Khan IA, Mehta RN, Sepkowitz DA. HIV-related pulmonary
hypertension: analytic review of 131 cases. Chest.2000; 118(4):1133
-1141.
PUBMED
36. Nunes H, Humbert M, Sitbon O, et al. Prognostic factors for
survival in human immunodeficiency virus-associated pulmonary arterial
hypertension. Am J Respir Crit Care Med.2003; 167(10):1433
-1439.
FREE FULL TEXT
37. Kanmogne GD, Primeaux C, Grammas P. Induction of apoptosis and
endothelin-1 secretion in primary human lung endothelial cells by HIV-1 gp120
proteins. Biochem Biophys Res Commun.2005; 333(4):1107
-1115.
PUBMED
38. Humbert M, Monti G, Fartoukh M, et al. Platelet-derived growth
factor expression in primary pulmonary hypertension: comparison of HIV
seropositive and HIV seronegative patients. Eur Respir
J. 1998;11
(3): 554-559.
ABSTRACT
39. Mette SA, Palevsky HI, Pietra GG, et al. Primary pulmonary
hypertension in association with human immunodeficiency virus infection: a
possible viral etiology for some forms of hypertensive pulmonary arteriopathy.
Am Rev Respir Dis.1992; 145(5):1196
-1200.
PUBMED
40. Marecki JC, Cool CD, Parr JE, et al. HIV-1 Nef is associated with
complex pulmonary vascular lesions in SHIV-Nef-infected macaques.
Am J Respir Crit Care Med.2006; 174(4):437
-445.
FREE FULL TEXT
41. Marecki JC, Cool CD, Beckey VE, Voelkel NF, Flores SC. HIV-1 Nef
protein is present a the site of plexiform lesions in paitients with
HIV-related pulmonary hypertension, and induces a program of altered
endothelial cell growth and survival in vitro. Proc Am Thorac
Soc. 2006;3:A476
.
42. Fauci AS, Pantaleo G, Stanley S, Weissman D. Immunopathogenic
mechanisms of HIV infection. Ann Intern Med.1996; 124(7):654
-663.
FREE FULL TEXT
43. Morse JH, Barst RJ, Itescu S, et al. Primary pulmonary hypertension
in HIV infection: an outcome determined by particular HLA class II alleles.
Am J Respir Crit Care Med.1996; 153(4 pt 1):1299
-1301.
FREE FULL TEXT
44. Ascherl G, Hohenadl C, Schatz O, et al. Infection with human
immunodeficiency virus-1 increases expression of vascular endothelial cell
growth factor in T cells: implications for acquired immunodeficiency
syndrome-associated vasculopathy. Blood.1999; 93 (12):4232
-4241.
FREE FULL TEXT
45. Caldwell RL, Gadipatti R, Lane KB, Shepherd VL. HIV-1 TAT represses
transcription of the bone morphogenic protein receptor-2 in U937 monocytic
cells. J Leukoc Biol.2006; 79(1):192
-201.
FREE FULL TEXT
46. Cool CD, Rai PR, Yeager ME, et al. Expression of human herpesvirus
8 in primary pulmonary hypertension. N Engl J Med.2003; 349(12):1113
-1122.
PUBMED
47. Nicastri E, Vizza CD, Carletti F, et al. Human herpesvirus 8 and
pulmonary hypertension. Emerg Infect Dis.2005; 11(9):1480
-1482.
PUBMED
48. Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV. Medical
therapy for pulmonary arterial hypertension: updated ACCP evidence-based
clinical practice guidelines. Chest.2007; 131(6):1917
-1928.
PUBMED
49. Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by
transfusions in children with sickle cell anemia and abnormal results on
transcranial Doppler ultrasonography. N Engl J Med.1998; 339(1):5
-11.
PUBMED
50. Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on
mortality and morbidity in adult sickle cell anemia: risks and benefits up to
9 years of treatment. JAMA.2003; 289(13):1645
-1651.
FREE FULL TEXT
51. Aessopos A, Farmakis D, Hatziliami A, et al. Cardiac status in
well-treated patients with thalassemia major. Eur J
Haematol.2004; 73(5):359
-366.
PUBMED
52. Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin
limits nitric oxide bioavailability in sickle-cell disease. Nat
Med.2002; 8(12):1383
-1389.
PUBMED
53. Channick RN, Newhart JW, Johnson FW, et al. Pulsed delivery of
inhaled nitric oxide to patients with primary pulmonary hypertension: an
ambulatory delivery system and initial clinical tests.
Chest. 1996;109
(6):1545
-1549.
PUBMED
54. Morris CR, Morris SM Jr, Hagar W, et al. Arginine therapy: a new
treatment for pulmonary hypertension in sickle cell disease? Am J
Respir Crit Care Med.2003; 168(1):63
-69.
FREE FULL TEXT
55. Derchi G, Forni GL, Formisano F, et al. Efficacy and safety of
sildenafil in the treatment of severe pulmonary hypertension in patients with
hemoglobinopathies. Haematologica.2005; 90(4):452
-458.
FREE FULL TEXT
56. Leong MH, Farber HW. Noninfectious pulmonary complications of HIV.
Clin Pulmonary Med. 2006;13
(3):194
-202.
ARTICLE EN RAPPORT
Cette semaine dans le JAMA
JAMA. 2008;299:253.
Texte Complet
|