|
|
Actions régulatrices pour la sécurité des produits biologiques approuvés aux Etats-Unis et dans l’Union européenne
Thijs J. Giezen, PharmD;
Aukje K. Mantel-Teeuwisse, PhD;
Sabine M. J. M. Straus, MD, PhD;
Huub Schellekens, PhD;
Antoine C. G. Egberts, PhD
JAMA. 2008;300(16):1887-1896
RÉSUMÉ
| |
Contexte Les produits biologiques représentent une classe nouvelle de médicaments porteurs de risques spécifiques (ex, immunogénicité). Cependant, une information limitée est disponible sur la nature et le moment des problèmes de sécurité concernant leur usage qui ont été identifiés après agrément.
Objectif Déterminer la nature, la fréquence, et le moment d’actions régulatrices de sécurité pour les produits biologiques suivant l’agrément aux Etats-Unis et/ou dans l’Union européenne.
Schéma et environnement Suivi d’un groupe de produits biologiques approuvés aux Etats-Unis et/ou dans l’Union européenne entre janvier 1995 et juin 2007. Les vaccins, les produits allergènes, et les produits pour une fabrication ultérieure et à des fins de transfusion ont été exclus.
Principaux critères de jugement La nature, la fréquence, et le moment des actions régulatrices de sécurité définies comme (1) des lettres aux prescripteurs (Etats-Unis) et des communications directes aux professionnels de santé (Union européenne), (2) avertissements black box (Etats-Unis), et (3) des retraits de commercialisation liés à la sécurité (Etats-Unis et Union Européenne) publiés entre janvier 1995 et juin 2008.
Résultats Un total de 174 produits biologiques a été approuvée (136 aux Etats-Unis et 105 dans l’Union européenne, dont 67 étaient approuvés dans les deux régions). Quatre-vingt deux actions régulatrices de sécurité (46 lettres aux prescripteurs, 17 communications directes aux professionnels de santé, 19 avertissements black box, et aucun retrait) ont été publiés pour 41 des 174 produits biologiques différents (23,6%). La probabilité d’une première action régulatrice de sécurité, dérivée d’analyses de Kaplan-Meier, était de 14% (intervalle de confiance 95% [IC], 9%-19%) 3 ans après agrément et 29% (IC 95%, 20%-37%) 10 ans après agrément. Les produits biologiques premiers dans une catégorie à obtenir un agrément présentaient un risque plus élevé d’action régulatrice de sécurité comparés aux produits approuvés ultérieurement dans cette même classe (12,0/1000 vs 2,9/1000 mois, respectivement ; rapport de hasard, 3,7 [IC 95%, 1,5-9,5]). Les avertissements concernaient les classes de pathologies générales et les conditions du site d’administration, les infections et infestations, les pathologies du système immunitaire et les néoplasmes bénins, malins et non spécifiés.
Conclusions La nature des problèmes de sécurité identifiés après agrément pour les produits biologiques est souvent liée à l’effet immuno-modulateur (infections). Du fait que les produits biologiques qui sont les premiers à être approuvés dans une classe étaient plus susceptibles d’être sujets à une action régulatrice, un contrôle serré est recommandé.
Les produits biologiques, définis comme produits dont la substance active est produite par ou extraite d’une source biologique, représentent une partie importante et croissante de l’arsenal thérapeutique.1 Aux Etats-Unis, le premier produit biologique, l’insuline recombinante, a été approuvé en octobre 1982.2 Depuis lors, plus de 250 produits biologiques, parmi lesquels les substances (sang) recombinantes, les produits monoclonaux basés sur des anticorps, et les vaccins recombinants ont été approuvés par les autorités de régulation.3 Entre 2003 et 2006, les produits biologiques représentaient 24% et 22% de toutes les nouvelles entités chimiques approuvées par les autorités de régulation des Etats-Unis et de l’Union Européenne.4 Les ventes de produits Biotech aux Etats-Unis ont montré un taux annuel croissant de 20% entre 2001 et 2006 comparé au taux de 6 à 8% sur le marché pharmaceutique.5
La connaissance d’un nouveau médicament est incomplète au moment de l’agrément, spécialement son profil de sécurité, du fait d’une variété de facteurs incluant des contraintes dans la taille de l’échantillon et la conception d’essais randomisés.6,7 Bien que ceci s’applique aussi à de petites molécules, les produits biologiques sont porteurs de risques spécifiques. Par contraste avec les petites molécules, qui sont synthétisées chimiquement, les produits biologiques sont dérivés de sources vivantes (ex, humains, animaux, cellules, et micro organismes). Le processus de production et de purification de produits biologiques est plus complexe, impliquant de nombreuses étapes avec le risque d’influencer les caractéristiques du produit terminal à chaque étape de la cascade de production.8,9 De petites différences et des changements dans le processus de production peuvent par conséquent avoir des implications majeures sur le profil sécurité des produits biologiques. Par exemple, l’incidence d’une érythroblastopénie chronique acquise chez les patients traités avec l’époétine humaine recombinante, une complication extrêmement rare induite par des anticorps, a été élevée chez les patients prenant une formulation particulière d’époétine humaine recombinante chez laquelle l’albumine sérique a été remplacée par du polysorbate 80 et la glycine.10,11 Cependant, le mécanisme exact sous-jacent le risque accru d’érythroblastopénie chronique acquise après le changement de formulation n’est pas encore totalement compris.12 Le risque de contamination avec des pathogènes par le donneur constitue un autre problème lié au processus de production (ex, pour les produits extraits du sang humain ou du plasma).13
Les produits biologiques sont spécifiquement enclins à l’induction de l’immunogénicité. Dans de nombreux cas, la conséquence de l’immunogénicité n’est pas adaptée cliniquement. Cependant, dans certains cas l’immunogénicité peut conduire à une perte d’efficacité du médicament ou, même pire, à une autoimmunité aux molécules endogènes. Il peut y avoir un impact clinique majeur si une protéine naturelle avec une activité biologique essentielle est neutralisée par une formation d’anticorps.8,10,14,15
La prévisibilité des données précliniques aux humains est limitée aux substances biologiques du fait de l’action spécifique aux espèces et aux propriétés immunogéniques chez les animaux.9 Un exemple frappant récent de cela a été la présence d’effets indésirables sérieux chez des volontaires sains participant à une étude clinique de phase 1 de TeGenero TGN1412, un anticorps agoniste CD 28. La tempête de cytokine observée suite à l’injection n’a pas été observée dans les études précliniques de TGN1412.16 Pour obtenir des résultats de valeur des études précliniques (toxicologie), un test animal pertinent devrait non seulement être sélectionné en se basant sur une activité pharmacologique et une basse immunogénicité, mais des propriétés pharmacocinétiques appropriées devraient aussi être prises en compte.17,18 Dans certains cas, le programme préclinique se complique davantage par une relation pharmacodynamique-pharmacocinétique qui peut être illustrée par l’effet retard pharmacodynamique du peg-interféron interleukine 2, qui devient apparent longtemps après que le médicament a disparu du compartiment sanguin. Un autre facteur de complication se trouve dans la présence de courbes de réponse en cloche, spécialement pour les cytokines, pour lesquelles l’effet désiré disparaît après une augmentation de la dose.19 Les produits biologiques agissent principalement extra-cellulairement et la toxicité est souvent attribuée à une pharmacologie exagérée,18 ce qui peut être illustré par la présence d’infections sérieuses dues à la fonction immuno-modulatoire de plusieurs substances biologiques (ex anticorps monoclonaux et interférons).20-22
Comme indiqué dans des études précédentes, l’utilisation de médicaments dans le cadre du monde réel de post-agrément peut conduire à l’identification de problèmes de sécurité importants, qui peut même résulter en un retrait de médicaments du marché.23,24 Du fait que les produits biologiques sont porteurs de risques spécifiques, et qu’une information limitée est disponible sur la nature et le chronométrage des actions de régulation liées à la sécurité publiées après agrément des produits biologiques, notre étude examine la nature des actions régulatrices de sécurité qui sont publiées après agrément. En se basant sur la fonction immuno-modulatoire de plusieurs produits biologiques, il est attendu qu’une part importante des avertissements à l’encontre des produits biologiques est liée à cette caractéristique. De plus, à l’intérieur du groupe de produits biologiques, les différences dans les actions régulatrices de sécurité (ex entre les classes mécaniques) sont aussi étudiées.
METHODES
Produits biologiques
Cette étude inclut des produits biologiques médicaux approuvés aux Etats-Unis et/ou dans l’Union européenne entre janvier 1995 et juin 2007. Les produits biologiques étaient définis en accord avec l’Agence européenne pour l’Evaluation des Médicaments (EMEA).1 Les substances actives identiques commercialisées par différentes compagnies pharmaceutiques et biosimilaires étaient comprises dans l’étude comme substances biologiques séparées.
Pour les Etats-Unis, l’étude comprenait des produits biologiques approuvées par le Centre d’Evaluation et de Recherche sur les Produits Biologiques (CBER) et le Centre d’Evaluation et de Recherche sur les Médicaments (CDER ; données disponibles depuis Janvier 1996). L’information a été obtenue à partir des sites web du CBER (http://www.fda.gov/cber) et CDER (http://www.accessdata.fda.gov/scipts/cder/drugsatfda), respectivement. Les produits biologiques agréés sous le même numéro de licence étaient inclus une seule fois. A l’intérieur de l’Union européenne, les produits biologiques qui ont reçu une autorisation de mise sur le marché ont été identifiés par les rapports d’évaluation publique européenne pour les produits médicinaux autorisés à usage humain. Toutes les informations étaient tirées du site Web de l’EMEA (http://www.emea.europa.eu).
Les produits biologiques avec une extension d’indication pendant la durée de l’étude, les vaccins, les produits allergéniques (tests épicutanés aux allergènes et extraits allergéniques), les produits biologiques pour une fabrication ultérieure, et les produits biologiques à des fins de transfusion et pour maintenir le volume de sang circulant étaient exclus.
Les produits biologiques étaient classés en catégories thérapeutiques selon le système de classification Chimique Thérapeutique Anatomique (http://www.whocc.no/atcddd), et étaient classifiés dans les classes d’anticorps (y compris les anticorps monoclonaux), les cytokines, les enzymes, les facteurs de croissance, les hormones, les interférons, les récepteurs, et autres/variés. Les anticorps monoclonaux étaient classifiés plus loin en murins, chimériques et humanisés.25,26
Actions régulatrices de sécurité
Les actions régulatrices de sécurité étaient définies comme (1) des communications écrites aux professionnels de santé (lettres aux prescripteurs [DH-PLs] aux Etats-Unis et des communications directes aux professionnels de santé [DHPCs] dans l’Union européenne), (2) des avertissements black box post agrément (aux Etats-Unis uniquement), et (3) des retraits du marché pour des raisons de sécurité (Etats-Unis et Union européenne). Des actions régulatrices de sécurité ont été collectées entre janvier 1995 et juin 2008, qui a assuré au moins 1 an de suivi pour chaque produit biologique à l’étude.
Les lettres aux prescripteurs étaient identifiées par MEDWATCH depuis 1996 (http://www.fda.gov/medwatch). Les communications directes aux professionnels de santé étaient identifiées à partir du site Web du Comité d’Evaluation des Médicaments aux Pays-Bas (http://www.cbg-mab.nl) et les rapports d’évaluation publique européenne de l’EMEA. La date de la lettre a été utilisée comme date d’action régulatrice de sécurité. Les lettres n’incluant pas des avertissements de sécurité et les lettres de suivi des lettres publiées précédemment ne contenant aucune information de sécurité ont été exclues.
Les avertissements black box (avertissement existant aux USA, figuré par un encadré noir placé au début de la notice, et signalant que le médicament en question présente des dangers pour la santé et des d’effets secondaires graves) étaient identifiés à partir des labels disponibles sur les sites Web de CDER, CBER, MEDWATCH, et le détenteur d’autorisation de mise sur le marché et les annonces mises sur le site Web de MEDWATCH. On a cherché l’avertissement black box pour le dernier label approuvé de chaque produit biologique disponible à partir de CDER. Lorsqu’un avertissement black box était identifié à partir du dernier label de garantie, on vérifiait des labels approuvés antérieurs pour identifier la date à laquelle l’avertissement black box était ajouté, ce qui était vérifié par recoupement avec l’information à partir de MEDWATCH. Les labels qui ne pouvaient pas être retirés de CDER étaient retirés de CBER, MEDWATCH, et/ou le détenteur de l’autorisation de mise sur le marché. La date de l’avertissement black box mentionnée sur les sites Web de CDER, CBER, ou MEDWATCH était incluse dans l’analyse. Lorsque la date exacte de l’avertissement black box ne pouvait pas être identifiée, la dernière date possible qui était publiée était incluse dans l’analyse. Aucun label ne pouvait être retiré pour un produit biologique, et ce produit biologique était exclu de l’analyse des avertissements black box.
Les retraits de médicaments pour des raisons de sécurité étaient identifiés à partir de MEDWATCH, CDER, et les rapports publics européens énumérés sur le site Web de l’EMEA. La date de la décision a été utilisée dans l’analyse.
Source et nature de l’information sur la sécurité
La source de l’action régulatrice de sécurité dans les communications aux professionnels de santé était collectée et classifiée comme rapports de post-agrément (y compris les deux rapports spontanés aussi bien que les études et registres pharmaco-épidémiologiques), les données d’essai clinique, une combinaison de rapports post-agrément et des données d’essai clinique, autres, ou inconnus. Du fait que l’information sur la source de l’action régulatrice de sécurité n’est pas incluse normalement dans l’avertissement black box, ces avertissements étaient exclus de la partie de l’analyse où l’action régulatrice de sécurité était étudiée.
La nature de l’information sur la sécurité était codée selon le Dictionnaire Médical pour les Autorisations de Régulation version 9.1. Les raisons primaires de la dissémination de l’action régulatrice de sécurité étaient incluses dans l’analyse. L’information de sécurité était encodée en utilisant 5 niveaux : le terme de niveau inférieur, le terme préféré, le terme de niveau plus élevé, le terme de groupe élevé, et la classe d’organe de système, mais seule la classe d’organe de système était utilisée dans l’analyse.
Analyse des données
Le nombre de produits biologiques classifiés au niveau Anatomique Chimique Thérapeutique aux Etats-Unis et dans l’Union européenne étaient comparés par le test du  2. Le délai moyen pour une action régulatrice de sécurité était calculé en additionnant les temps entre l’agrément et l’avertissement et en divisant par le nombre total d’actions régulatrices publiées. L’incidence des actions régulatrices était calculée comme une simple proportion. Les courbes de survie de Kaplan-Meier étaient utilisées pour estimer la probabilité d’occurrence d’action régulatrice de sécurité pour le groupe total de produits biologiques (ceux approuvés soit aux Etats-Unis soit dans l’Union européenne), et pour les sous-groupes (ceux approuvés dans l’Union européenne et aux Etats-Unis séparément, selon l’action régulatrice d’intérêt liée à la sécurité. Pour les produits biologiques qui ont acquis de multiples actions régulatrices de sécurité, seule la première action régulatrice était incluse dans l’analyse. 2. Le délai moyen pour une action régulatrice de sécurité était calculé en additionnant les temps entre l’agrément et l’avertissement et en divisant par le nombre total d’actions régulatrices publiées. L’incidence des actions régulatrices était calculée comme une simple proportion. Les courbes de survie de Kaplan-Meier étaient utilisées pour estimer la probabilité d’occurrence d’action régulatrice de sécurité pour le groupe total de produits biologiques (ceux approuvés soit aux Etats-Unis soit dans l’Union européenne), et pour les sous-groupes (ceux approuvés dans l’Union européenne et aux Etats-Unis séparément, selon l’action régulatrice d’intérêt liée à la sécurité. Pour les produits biologiques qui ont acquis de multiples actions régulatrices de sécurité, seule la première action régulatrice était incluse dans l’analyse.
Les rapports de hasard (HR) avec un intervalle de confiance de 95% (IC) étaient calculés en utilisant le modèle de risques proportionnels de Cox. Les HR étaient calculés pour évaluer si le premier produit biologique approuvé dans un (nouveau) sous-groupe chimique, pharmacologique, et thérapeutique, comme défini par le système de classification Anatomique Thérapeutique Chimique, présentait un risque plus élevé d’action régulatrice de sécurité comparé aux produits biologiques approuvés à un stade ultérieur.
Pour évaluer si l’expérimentation avec des produits biologiques (ceux approuvés à un stade ultérieur dans le temps) pouvait influencer le nombre d’actions régulatrices de sécurité, des cadres à 2 temps ont été construits qui incluaient des produits biologiques approuvés entre janvier 1995 et juin 2007 (6 ans). Les HR étaient aussi calculés pour comparer le risque d’actions régulatrices de sécurité publié pour les différentes classes mécaniques de produits biologiques décrits avant. Les hormones étaient utilisées comme groupe de référence parce qu’il existe une expérience à long terme, extensive avec les hormones à l’intérieur du groupe de produits biologiques, et les hormones sont souvent des imitations de substances qui apparaissent naturellement.26 Pour les sous-groupes de produits biologiques approuvés aux Etats-Unis et dans l’Union européenne, les distributions du temps écoulé jusqu’à l’événement des DHPL publiées aux Etats-Unis et les DHPC publiées dans l’Union européenne étaient comparées par les HR, qui incluaient un ajustement de la variance pour représenter une indépendance statistique.
Pour un sous-groupe de produits biologiques à la fois aux Etats-Unis et dans l’Union européenne, la nature et le moment choisi de l’action régulatrice de sécurité étaient comparées de façon descriptive. Lorsqu’une action régulatrice de sécurité était publiée pour un produit biologique non approuvé dans une autre région au moment de l’action régulatrice, cette action régulatrice était exclue. Le moment choisi d’un avertissement régulateur de sécurité était classifié (1) d’abord aux Etats-Unis (dans l’Union européenne >2 mois plus tard), (2) dans l’Union européenne d’abord (aux Etats-Unis >2 mois plus tard), et (3) à la fois aux Etats-Unis et dans l’Union européenne dans une période de 2 mois.
Toutes les analyses statistiques étaient menées en utilisant le package de logiciel SPSS version 14 (SPSS Inc., Chicago, Illinois) et la version 6.2 S-PLUS (Insightful Corp, Seattle, Washington). Toutes les hypothèses étaient testées en utilisant des tests de corrélation avec un niveau  de .05. Toutes les analyses n’étaient pas ajustées parce que l’objectif de cette étude était descriptif et non étiologique par nature. de .05. Toutes les analyses n’étaient pas ajustées parce que l’objectif de cette étude était descriptif et non étiologique par nature.
RESULTATS
Un total de 174 produits biologiques médicaux ont obtenu un agrément entre janvier 1995 et juin 2007 ; ceci comprenait 136 produits biologiques approuvés aux Etats-Unis et 105 dans l’Union européenne, parmi lesquels 67 produits biologiques ont obtenu un agrément dans les deux zones à la fois pendant la durée de l’étude (TABLEAU 1).
|
|
|
|
Tableau 1. Produits biologiques approuvés entre janvier 1995 et juin 2007 et classifiés en catégorie thérapeutique (N = 174)
|
|
|
Les différences entre le nombre de produits biologiques approuvés aux Etats-Unis et dans l’Union européenne était surtout expliquées par les différences dans les classes Anatomiques Thérapeutiques Chimiques d’insulines et analogues (P<.001), d’autres préparations anti-anémiques (P = 0.10), des hormones du lobe antérieur de l’hypophyse et analogues (P = .08) et les immunoglobulines (P =0.01).
Pendant la période examinée, 82 actions régulatrices de sécurité ont été publiées pour 41 des 174 produits biologiques (23,6%). Ceux-ci incluaient 46 DHPL (TABLEAU 2), 17 DHPC (TABLEAU 3) et 19 avertissements black box (TABLEAU 4). Aucun produit biologique n’a été retiré pour des raisons de sécurité. Le temps moyen pour mettre à jour une action régulatrice de sécurité était de 3,7 ans et 70,7% des actions régulatrices étaient publiées dans l’intervalle de 5 ans après agrément. La probabilité de Kaplan-Meier d’un produit biologique nécessitant sa première action régulatrice de sécurité était de 14% (IC 95%, 9%-19%) 3 ans après agrément et 29% (IC 95%, 20%-37%) 10 ans après agrément.
|
|
|
|
Tableau 2. Produits biologiques avec une lettre aux prescripteurs (DHPL) aux Etats-Unis
|
|
|
|
|
|
|
Tableau 3. Produits biologiques avec une communication directe aux professionnels de santé (DHPC) dans l’Union européenne
|
|
|
|
|
|
|
Tableau 4. Produits biologiques avec un avertissement black box (BBW) aux Etats-Unis
|
|
|
Les produits biologiques qui étaient les premiers à être approuvés dans leur sous-groupe chimique, pharmacologique et thérapeutique, présentaient un risque significativement plus élevé pour l’occurrence de sa première action régulatrice que des produits approuvés plus tard (RR, 3,7 ; IC 95%, 1,5-9,5). Lorsque les premiers produits incluaient aussi des produits biologiques approuvés dans un sous-groupe chimique, pharmacologique et thérapeutique, dans lesquels les petites molécules avaient été approuvées, un risque significativement accru pour la première action régulatrice avait été trouvé également (RR, 2,3 ; IC 95%, 1,1-4,8) (TABLEAU 5). Les produits biologiques approuvés entre juillet 2001 et juin 2007 présentaient un risque non significativement plus élevé pour leur action régulatrice de sécurité comparé aux produits biologiques approuvés entre janvier 1995 et juin 2001 (RR, 1,5 ; IC 95%, 0,8-2,8) (TABLEAU 5).
|
|
|
|
Tableau 5. Risques pour une action régulatrice de sécurité
|
|
|
Un risque significativement plus élevé pour une action régulatrice comparée aux hormones a été trouvé pour les anticorps (RR, 12,1 IC 95%, 3,5-86,1), les facteurs de croissance (RR, 8,2 ; IC 95%, 1,4-49,1), les interférons (RR, 7,3 ; IC 95%, 1,6-32,8) et les récepteurs (RR, 34,2 ; IC 95%, 5,6-211,1). Comparés aux anticorps monoclonaux humanisés, les anticorps monoclonaux chimériques présentaient un risque non significativement plus élevé (RR, 2,9 ; IC 95%, 0,9-9,9) et les anticorps monoclonaux murins présentaient un risque plus bas (RR, 0,2 ; IC 95%, 0,03-0,7) pour une première action régulatrice de sécurité (TABLEAU 5).
Les actions régulatrices de sécurité publiées pour les produits biologiques concernaient principalement les classes d’organes de système de pathologies générales et les conditions du site d’administration (26,8% sur 82), les infections et les infestations (22%), les troubles du système immunitaire (15,9%), et les néoplasmes bénins, malins et non spécifiés (12,2%) (TABLEAUX 2, 3, 4). Une description quantitative de la fréquence de chaque type d’action régulatrice de sécurité est montrée TABLEAU 6.
|
|
|
|
Tableau 6. Actions régulatrices de sécurité classifiées au niveau de système classe organea
|
|
|
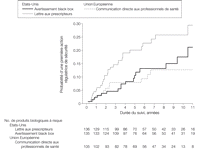
Les 46 DHPL, 17 DHPC et 19 avertissements black box de sécurité étaient publiés pour 30 (22,1%), 11 (10 ?5%), et 17 (12,6%) différents produits biologiques, respectivement. Après 10 ans, la probabilité d’un premier DHPL était de 26% (IC 95%, 17%-34%) ; DHPC, 13% (IC 95%, 5%-20%) ; et avertissement black box, 17% (IC 95%, 8%-26%) (FIGURE). Les communications aux professionnels de santé étaient publiées plus fréquemment aux Etats-Unis que dans l’Union européenne (RR, 3,0 ; IC 95%, 1,2-7,6).
|
|
|
Figure. Actions régulatrices de sécurité pour les produits biologiques
|
|
|
Les sources des actions régulatrices décrites dans les DHPL étaient des rapports de post-agrément (n = 18 ; 39,1%), des données d’essai clinique (n = 9 ; 19,6%), autres (n = 2 ; 4,3%) et inconnus (n = 1 ; 2,2%°. Les sources d’actions régulatrices de sécurité décrites dans les DHPC étaient des rapports post-agrément et des données cliniques (n = 1 ; 5,9%), autres (n = 1 ; 5,9%), et inconnus (n = 1 ; 5,9%).
Soixante-et-une actions régulatrices de sécurité étaient publiées pour le sous-groupe de 67 produits biologiques approuvés à la fois aux Etats-Unis et dans l’Union européenne à l’intérieur de la période étudiée. Cinq actions régulatrices de sécurité publiées aux Etats-Unis ont été exclues parce que le produit biologique n’était pas approuvé dans l’Union européenne à l’époque de l’action régulatrice. Neuf actions régulatrices de sécurité publiées dans les deux régions impliquaient la même nature (35 étaient publiées aux Etats-Unis seulement et 8 dans l’Union européenne uniquement). Des 9 actions régulatrices de sécurité publiées dans les deux régions, 6 étaient publiées dans les deux régions en 2 mois (1 était publiée d’abord aux Etats-Unis et 2 étaient publiées d’abord dans l’Union européenne).
COMMENTAIRE
L’expérience avec les médicaments en pratique clinique actuelle complète celle d’essais cliniques et aide à étendre la connaissance du profil de sécurité des médicaments.7,23,24 Les produits biologiques représentent une classe relativement nouvelle de médicaments et les actions régulatrices de sécurité publiées dans la classe d’organe de système des infections et des infestations touchant l’effet immuno-modulateur de nombreux produits biologiques a été confirmé par la présente étude. Les actions régulatrices de sécurité publiées dans la classe d’organe de système des pathologies générales et les conditions du site d’administration peuvent être expliquées en partie par les réactions aux injections qui se produisent après le trajet d’administration parentéral, qui est le mode d’administration de la plupart des produits biologiques. Une évaluation plus en profondeur du mode d’administration des produits biologiques pourrait avoir prédit quelques problèmes de sécurité pendant la phase de développement.
Le facteur de nécrose tumorale, par exemple, est libéré par des macrophages activés, les lymphocytes T, et d’autres cellules immunes et joue un rôle important dans la réponse immune humaine aux infections.27,28 Comme le montre l‘étude présente, le risque d’infections avec l’anticorps infliximab du facteur de nécrose tumorale a été identifié et communiqué après agrément du médicament. L’association entre des anti-inflammatoires non stéroïdiens et l’occurrence d’événements indésirables du tractus gastro-intestinal, illustre le besoin d’une évaluation en profondeur du mode d’action est valable aussi pour les petites molécules.29
Il existe des données limitées disponibles sur le timing et la fréquence des actions régulatrices de sécurité publiées en post-agrément. L’étude présente a montré que les 174 produits biologiques avaient une probabilité de 14% pour nécessiter leur première action régulatrice de sécurité dans les 3 ans après avoir reçu l’autorisation de mise sur le marché, il est important de noter que tous les médicaments ne sont pas commercialisés immédiatement après agrément et certains médicaments ne seront jamais commercialisés. Du fait que tous les produits biologiques qui avaient obtenu l’autorisation de mise sur le marché étaient inclus dans l’étude actuelle, ceci peut avoir conduit à une sous-estimation de la probabilité d’une action régulatrice de sécurité, qui est une limitation à cette étude. Malheureusement, le statut commercial ne pouvait être retiré des sites Web de la Food and Drug Administration et de l’EMEA.
Lasser et al23 a trouvé que de nouvelles entités chimiques approuvées jusqu’en 1999 avait une probabilité d’avertissement black box de 10% après 10 ans de commercialisation comparé aux 17% trouvés dans notre étude. Les produits biologiques, par conséquent, semblent être plus susceptibles à un avertissement black box publié en post-agrément comparé aux nouvelles entités chimiques en général. Néanmoins, des différences existant entre les deux études, avec l’inclusion de multiples avertissements black box publiés pour le même médicament et un retard dans l’occurrence de l’avertissement black box dans le Physicians’ Desk Reference par Lasser et al23,30 et une conscience accrue de la sécurité du patient et de l’accessibilité aux données de sécurité dans le temps,31 elles écartent une comparaison directe des probabilités.
Une limitation qui devrait être abordée est la taille relativement petite de l’échantillon et le petit nombre d’actions régulatrices de sécurité résultant en de larges IC de 95%, ce qui rend l’interprétation des découvertes non statistiquement significative stimulante. Cependant, il a été décidé de n’inclure que les produits biologiques à partir de 1995 parce qu’une prise de décision centralisée par l’EMEA pour tous les produits biologiques a commencé en 1995.
Comme on le voit dans cette étude, la décision pour une action régulatrice de sécurité peut être basée sur des essais (larges) cliniques, des rapports de cas, et/ou des études épidémiologiques et peuvent ainsi être basées sur des observations cliniques sans aucune confirmation épidémiologique ou expérimentale. De plus, le nombre de patients exposés aux différents médicaments peut varier énormément, ce qui peut affecter à la fois la détection du problème de sécurité aussi bien que sa signification pour la pratique clinique. Ce dilemme dans la prise de décisions — disponibilité de données incomplètes et variabilité dans l’exposition du patient — est reconnu. Cependant la dissémination d’une action régulatrice est habituellement le résultat d’une évaluation équilibrée par les autorités régulatrices prenant en compte le sérieux du problème de sécurité et le besoin d’informer les professionnels de santé. De vastes études épidémiologiques et/ou des essais cliniques sont souvent nécessaires pour confirmer l’association entre le problème de sécurité résultant en une action régulatrice et le médicament. L’effet des études épidémiologiques pour l’identification des problèmes de sécurité ne pouvait pas être pris en compte dans cette étude parce qu’il n’a pas été possible de faire la différence entre les problèmes de sécurité identifiés par des rapports spontanés et les résultats d’études épidémiologiques basés sur les communications aux professionnels de santé.
Certains des produits biologiques (par exemple, les anticorps monoclonaux) diffèrent essentiellement de substances existant naturellement et peuvent par conséquent être spécialement susceptibles aux réactions de médicaments indésirables.26 Bien que les IC de 95% étaient larges, notre étude a confirmé que ceux-ci et d’autres produits biologiques, y compris des cytokines, des facteurs de croissance, des interférons, et des récepteurs étaient spécifiquement enclins à des actions régulatrices de sécurité. A l’intérieur du groupe des anticorps monoclonaux, les anticorps murins avaient un risque plus bas pour une action comparés aux anticorps monoclonaux humanisés. Cependant, cette découverte devrait être interprétée avec prudence du fait du petit nombre d’anticorps monoclonaux et d’actions régulatrices de sécurité.
Les premiers produits biologiques approuvés dans un sous-groupe chimique, pharmacologique, et thérapeutique étaient à un risque plus élevé pour leur première action régulatrice de sécurité. Cette découverte suggère que la pharmacovigilance devrait être spécialement rigoureuse pour les premiers produits biologiques pour être approuvés dans un sous-groupe chimique, pharmacologique et thérapeutique. Notre étude a aussi montré que les produits biologiques approuvés les 6 dernières années de la période d’étude (de juillet 2001 à juin 2007) avait un risque significatif plus élevé pour leur première action régulatrice de sécurité comparé avec les produits biologiques approuvés pendant les 6,5 premières années de la durée de l’étude (janvier 1995-juin 2001). Ce risque plus élevé est principalement dû au nombre élevé de DHPL publié en 2005. La plupart des produits biologiques approuvés dans un nouveau sous-groupe chimique, pharmacologique, et thérapeutique était approuvée pendant les 6,5 premières années de la durée de l’étude.
Des différences existent dans la nature des actions régulatrices de sécurité pour les produits biologiques comparées aux petites molécules. Comme on le sait à partir d’études précédentes, la plupart des problèmes de sécurité identifiés en post-agrément pour les petites molécules sont identifiés dans le système de classe organe des pathologies hépatobiliaires, pathologies du sang et du système lymphatique, pathologies cardiaques, et pathologies du système nerveux.23,24,32 La connaissance de la nature des événements de sécurité liée aux produits biologiques et la différence entre les petites molécules et les produits biologiques relativement nouveaux semble pertinente. Le manque de conscience de la nature des problèmes de sécurité liés aux produits biologiques pourrait gêner le lien entre le produit biologique et son effet indésirable lorsqu’un patient présente un problème clinique.
La plupart des lettres étaient disséminées aux Etats-Unis comparées à l’Union européenne, ce qui correspond à une observation précédente disant que l’approche d’une information de sécurité apparaissait comme étant plus conservatrice dans le résumé des caractéristiques des produits de l’UE que dans l’encart du package US.33 Dans les deux régions, la dissémination d’une lettre peut être initiée par le détenteur de l’autorisation de mise sur le marché lorsqu’une information importante de sécurité est identifiée et lorsque des changements importants ont été faits pour l’étiquetage du produit.34,35 De plus, aux Etats-Unis une lettre peut être publiée pour souligner des corrections à une publicité de médicament sous-prescription ou à un étiquetage aussi.
Seuls 67 produits biologiques ont été approuvés dans les deux régions pendant la durée de l’étude. On devrait noter que certains produits ont pu être approuvés dans l’autre région avant le moment de l’étude. Des 56 actions régulatrices de sécurité publiées pour ces 67 produits biologiques, seuls 9 actions régulatrices de sécurité impliquaient le même type d’avertissement de sécurité. Ceci semble pertinent parce que les 35 autres et les 8 actions régulatrices de sécurité publiées aux Etats-Unis et dans l’Union européenne, respectivement, n’étaient pas publiées dans l’autre région. Du fait que seul le résumé approuvé le plus récent des caractéristiques des produits était disponible à partir du site Web EMEA, il n’a pas été possible de chercher si les effets indésirables des médicaments communiqués par une DHPL aux Etats-Unis étaient déjà inclus dans le résumé de l’UE des caractéristiques du produit sans communication par une DHPC. Six des 9 avertissements publiés dans les deux régions étaient publiés dans un laps de temps de 2 mois.
En résumé, cette étude a montré que presque 50% des actions régulatrices de sécurité pour les produits biologiques étaient publiées dans le système classe organe des pathologies générales et des sites d’administration, d’infections et d’infestations. Les avertissements publiés dans le système classe organe d’infections et d’infestations étaient souvent liés à l’effet immuno-modulateur de nombreux produits biologiques. Bien que les limitations des essais précliniques pour les produits biologiques soient reconnues, les résultats d’études pharmacologiques, d’études précliniques et d’études cliniques pourraient résulter en la prédiction de risques potentiels liés au médicament pour lequel un contrôle serré est requis dans le cadre de post-agrément. Les professionnels de santé devraient être conscients du risque spécifique lié à la relativement nouvelle classe de produits biologiques pour pouvoir fournir un lien entre l’utilisation de produit biologique et le patient présentant un problème clinique. De plus, les classes d’anticorps, (monoclonaux), les cytokines, les facteurs de croissance, les interférons, et les récepteurs et les premiers produits biologiques approuvés dans un sous-groupe chimique, pharmacologique et thérapeutique sont spécifiquement enclins à une première action régulatrice de sécurité ; un contrôle serré de ces produits biologiques est par conséquent recommandé.
Informations sur les auteurs
Correspondance: Aukje K. Mantel-Teeuwisse, PhD, Utrecht Institute for Pharmaceutical Sciences, PO Box 80082, 3508 TB Utrecht, Pays-Bas (A.K.Mantel{at}uu.nl).
Contributions de l’auteur : Le Dr Giezen a eu un accès complet à toutes les données de l’étude et accepte la responsabilité de l’intégrité des données et de l’exactitude de l’analyse des données.
Concept de l’étude et conception : Giezen, Mantel-Teeuwisse, Straus, Leufkens, Egberts.
Acquisition des données : Giezen, Mantel-Teeuwisse.
Analyse et interprétation des données : Giezen, Mantel-Teeuwisse, Schellekens, Leufkens, Egberts.
Elaboration du manuscrit : Giezen, Mantel-Teeuwisse
Révision critique du manuscrit : Mantel-Teeuwisse, Straus, Schellekens, Leufkens, Egberts.
Analyse statistique : Giezen, Mantel-Teeuwisse, Egberts.
Soutien administratif, technique ou matériel : Giezen, Mantel-Teeuwisse, Egberts.
Surveillance de l’étude : Mantel-Teeuwisse, Straus, Schellekens, Leufkens, Egberts.
Liens financiers : Le département employant les Drs Giezen, Mantel-Teeuwisse, Leufkens, et Egberts a reçu des bourses de recherche sans restriction de GlaxoSmithKline, Organon, Merck, et Novo Nordisk pour la conduite de la recherche pharmacoépidémiologique. Le Dr Schellekens a rapporté avoir reçu des bourses de recherche de Organon, Roche, et Merck-Serono. Le Dr. Strauss n’a rapporté aucun lien financier.
Affiliations des auteurs: Utrecht Institute for Pharmaceutical Sciences, Divisions of Pharmacoepidemiology and Pharmacotherapy (Drs Giezen, Mantel-Teeuwisse, Leufkens, et Egberts) and Pharmaceutics (Dr Schellekens), and Department of Innovation Sciences (Dr Schellekens), Utrecht University, and Department of Clinical Pharmacy, University Medical Center Utrecht (Dr Egberts), Utrecht, Pays-Bas; Medicines Evaluation Board, the Hague, the Netherlands (Drs Giezen, Mantel-Teeuwisse, Straus, et Leufkens).
Pour les commentaires éditoriaux voir p 1939.
BIBLIOGRAPHIE
| |
1. Commission of European Communities. Commission Directive 2003/63/EC of 25 June 2003 amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use. http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-1/dir_2003_63/dir_2003_63_en.pdf. Accessibility verified September 11, 2008.
2. Frank RG. Regulation of follow-on biologics. N Engl J Med. 2007;357(9):841-843.
PUBMED
3. Shankar G, Pendley C, Stein KE. A risk-based bioanalytical strategy for the assessment of antibody immune responses against biological drugs. Nat Biotechnol. 2007;25(5):555-561.
PUBMED
4. Walsh G.. Biopharmaceutical benchmarks 2006. Nat Biotechnol. 2006;24(7):769-776.
PUBMED
5. Aggarwal S.. What’s fueling the biotech engine? Nat Biotechnol. 2007;25(10):1097-1104.
PUBMED
6. Schneeweiss S.. Developments in post-marketing comparative effectiveness research. Clin Pharmacol Ther. 2007;82(2):143-156.
PUBMED
7. Stricker BH, Psaty BM. Detection, verification, and quantification of adverse drug reactions. BMJ. 2004;329(7456):44-47.
FREE FULL TEXT
8. Schellekens H.. Follow-on biologics: challenges of the "next generation." Nephrol Dial Transplant. 2005;20(suppl 4):iv31-iv36.
ABSTRACT
9. Baumann A.. Early development of therapeutic biologics–pharmacokinetics. Curr Drug Metab. 2006;7(1):15-21.
PUBMED
10. Schellekens H.. Immunologic mechanisms of EPOassociated pure red cell aplasia. Best Pract Res Clin Haematol. 2005;18(3):473-480.
PUBMED
11. Schellekens H, Ryff JC. "Biogenerics": the offpatent biotech products. Trends Pharmacol Sci. 2002;23(3):119-121.
PUBMED
12. Schellekens H, Jiskoot W.. Eprex-associated pure red cell aplasia and leachates. Nat Biotechnol. 2006;24(6):613-614.
PUBMED
13. Vonberg RP, Gastmeier P. Hospital-acquired infections related to contaminated substances. J Hosp Infect. 2007;65(1):15-23.
PUBMED
14. Ryff JC, Schellekens H. Immunogenicity of rDNAderived pharmaceuticals. Trends Pharmacol Sci. 2002;23(6):254-256.
PUBMED
15. Kessler M, Goldsmith D, Schellekens H.. Immunogenicity of biopharmaceuticals. Nephrol Dial Transplant. 2006;21(suppl 5):v9-v12.
FREE FULL TEXT
16. Suntharalingam G, Perry MR, Ward S, et al.. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018-1028.
PUBMED
17. Sims J.. Assessment of biotechnology products for therapeutic use. Toxicol Lett. 2001;120(1-3):59-66.
PUBMED
18. Brennan FR, Shaw L, Wing MG, Robinson C. Preclinical safety testing of biotechnology-derived pharmaceuticals: understanding the issues and addressing the challenges. Mol Biotechnol. 2004;27(1):59-74.
PUBMED
19. Crommelin DJ, Storm G, Verrijk R, de Leede L, Jiskoot W, Hennink WE. Shifting paradigms: biopharmaceuticals versus low molecular weight drugs. Int J Pharm. 2003;266(1-2):3-16.
PUBMED
20. Hamilton CD. Infectious complications of treatment with biologic agents. Curr Opin Rheumatol. 2004;16(4):393-398.
PUBMED
21. Aksamit AJ. Review of progressive multifocal leukoencephalopathy and natalizumab. Neurologist. 2006;12(6):293-298.
PUBMED
22. Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295(19):2275-2285.
FREE FULL TEXT
23. Lasser KE, Allen PD, Woolhandler SJ, Himmelstein DU, Wolfe SM, Bor DH. Timing of new black box warnings and withdrawals for prescription medications. JAMA. 2002;287(17):2215-2220.
FREE FULL TEXT
24. Bakke OM, Manocchia M, de Abajo F, Kaitin KI, Lasagna L. Drug safety discontinuations in the United Kingdom, the United States, and Spain from 1974 through 1993: a regulatory perspective. Clin Pharmacol Ther. 1995;58(1):108-117.
PUBMED
25. Kromminga A, Schellekens H.. Antibodies against erythropoietin and other protein-based therapeutics: an overview. Ann N Y Acad Sci. 2005;1050:257-265.
PUBMED
26. Schellekens H, Bragt PH, Olijve W, Van der Weele CN. Medische Biotechnologie. Maarssen, the Netherlands: Elsevier Gezondheidszorg; 2001.27. Winthrop KL. Risk and prevention of tuberculosis and other serious opportunistic infections associated with the inhibition of tumor necrosis factor. Nat Clin Pract Rheumatol. 2006;2(11):602-610.
PUBMED
28. Botsios C.. Safety of tumour necrosis factor and interleukin-1 blocking agents in rheumatic diseases. Autoimmun Rev. 2005;4(3):162-170.
PUBMED
29. Brune K, Furst DE. Combining enzyme specificity and tissue selectivity of cyclooxygenase inhibitors: towards better tolerability? Rheumatology (Oxford). 2007;46(6):911-919.
PUBMED
30. Temple RJ, Himmel MH. Safety of newly approved drugs: implications for prescribing. JAMA. 2002;287(17):2273-2275.
FREE FULL TEXT
31. Hartford CG, Petchel KS, Mickail H, et al.. Pharmacovigilance during the pre-approval phases: an evolving pharmaceutical industry model in response to ICH E2E, CIOMS VI, FDA and EMEA/CHMP risk management guidelines. Drug Saf. 2006;29(8):657-673.
PUBMED
32. Meyboom RHB, Gribnau FWJ, Hekster YA, De Koning GHP, Egberts ACG. Characteristics of topics in pharmacovigilance in the Netherlands. Clin Drug Investig. 1996;12:207-219.
33. Nieminen O, Kurki P, Nordstrom K.. Differences in product information of biopharmaceuticals in the EU and the USA: implications for product development. Eur J Pharm Biopharm. 2005;60(3):319-326.
PUBMED
34. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research.. Drug safety information: FDA’s communication to the public, March 2007. http://www.fda.gov/cder/guidance/7477fnl.pdf. Accessibility verified September 10, 2008.35. European Commission. Volume 9A of the rules governing medicinal products in the European Union: guidelines on pharmacovigilance for medicinal products for human use, March 2007. http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-9/pdf/vol9A_2007-04.pdf. Accessibility verified September 10, 2008.
ARTICLES EN RAPPORT
Cette semaine dans le JAMA-Français
JAMA. 2008;300:1845.
Texte Complet
Médicaments sur prescription, responsabilité des produits, et préemption de responsabilité civile
Catherine D. DeAngelis et Phil B. Fontanarosa
JAMA. 2008;300:1939-1941.
Texte Complet
|