|
|
JAMA-EXPRESS
Effets de la ranolazine sur la récurrence des événements cardiovasculaires chez des patients avec syndromes coronariens aigus sans surélévation du segment STÉtude randomisée MERLIN-TIMI 36
David A. Morrow, MD, MPH;
Benjamin M. Scirica, MD, MPH;
Ewa Karwatowska-Prokopczuk, MD;
Sabina A. Murphy, MPH;
Andrzej Budaj, MD;
Sergei Varshavsky, MD;
Andrew A. Wolff, MD;
Allan Skene, PhD;
Carolyn H. McCabe, BS;
Eugene Braunwald, MD; Pour les investigateurs de l'étude MERLIN-TIMI 36
RÉSUMÉ
| |
Contexte La ranolazine est un nouvel antiangineux qui réduit
l'ischémie chez les patients avec angor chronique; cependant, elle n'a
pas été étudiée chez les patients souffrant de
syndromes coronariens aigus (SCA).
Objectif Déterminer l'efficacité et l'innocuité
de la ranolazine au cours du traitement à long terme des patients avec
SCA sans surélévation du segment ST.
Schéma, cadre et participants Étude internationale
randomisée en double aveugle, contrôlée contre placebo, de
6 560 patients dans les 48 heures suivant l'apparition de leurs
symptômes ischémiques, qui ont été traités
par ranolazine (initiation par voie intraveineuse, puis prise orale de
ranolazine à libération prolongée à la dose de 1
000 mg deux fois par jour, n = 3 279) ou par un placebo correspondant (n = 3
281), et suivis pendant une médiane de 348 jours dans l'étude
MERLIN (Metabolic Efficiency With Ranolazine for Less Ischemia in
Non-ST-Elevation Acute Coronary Syndromes)-TIMI 36, entre le 8 octobre 2004 et
le 14 février 2007.
Principaux critères d'évaluation Le critère
d'efficacité primaire était un critère composite incluant
le décès cardiovasculaire, l'infarctus du myocarde (IDM) ou
l'ischémie récurrente jusqu'à la fin de l'étude.
Les principaux critères d'innocuité étaient le
décès toutes causes confondues et l'arythmie symptomatique
documentée.
Résultats Le critère primaire est survenu chez 696
patients (21,8 %) dans le groupe ranolazine et chez 753 patients (23,5 %) dans
le groupe placebo (rapport des risques [HR], 0,92; intervalle de confiance
[IC] à 95 %, 0,83-1,02; p = 0,11). Le principal critère
secondaire (décès cardiovasculaire, IDM ou ischémie
sévère récurrente) est survenu chez 602 patients (18,7%)
du groupe ranolazine et chez 625 patients (19,2 %) du groupe placebo (HR,
0,96; IC 95 %, 0,86-1,08; p = 0,50). Le décès cardiovasculaire
ou IDM a été observé chez 338 patients (10,4 %)
assignés à la ranolazine et 343 patients (10,5 %)
assignés au placebo (HR, 0,99; IC 95 %, 0,85-1,15; p = 0,87).
L'ischémie récurrente était inférieure dans le
groupe ranolazine (430 [13,9 %]) comparé au groupe placebo (494 [16,1
%]; HR, 0,87; IC 95 %, 0,76-0,99; p = 0,03). L'allongement du QTc
nécessitant une réduction posologique du traitement intraveineux
a été observé chez 31 patients (0,9 %) recevant la
ranolazine comparé à 10 patients (0,3%) sous placebo. Les
arythmies symptomatiques documentées ne différaient pas entre
les groupes ranolazine (99 [3,0 %]) et placebo (102 [3,1 %]); (p = 0,84).
Aucune différence dans la mortalité totale n'a été
observée avec la ranolazine comparée au placebo (172 vs 175; HR,
0,99; IC 95 %, 0,80-1,22; p = 0,91).
Conclusions L'adjonction de la ranolazine au traitement de
référence des SCA n'a pas démontré
d'efficacité dans la réduction des événements
cardiovasculaires majeurs. La ranolazine n'a pas eu d'effet défavorable
sur le risque de décès toutes causes confondues ou d'arythmie
symptomatique documentée. Nos résultats soutiennent
l'innocuité et l'efficacité de la ranolazine dans le traitement
antiangineux.
JAMA.
2007;297:1775-1783
Le syndrome coronarien aigu (SCA) sans surélévation du
segment ST est une pathologie hétérogène, avec de
multiples étiologies possibles qui peuvent contribuer à un
déséquilibre entre les apports et les besoins myocardiques en
oxygène, résultant en une perturbation de l'homéostasie
cellulaire et en une déplétion des stocks
énergétiques des cellules
myocardiques.1
La prise en charge actuelle de ce syndrome en phase aiguë vise
essentiellement l'amélioration de l'apport en oxygène
myocardique par la réduction de la thrombose coronaire limitant le
flux, ainsi que la revascularisation de l'athérosclérose
oblitérante sousjacente; elle est en outre associée à des
interventions visant à réduire le besoin en oxygène
myocardique.
2 Le
traitement chronique a comme double objectif de prévenir la survenue de
nouveaux événements cardiovasculaires majeurs et de
réduire la récurrence des symptômes
ischémiques.3
Malgré les progrès réalisés dans le traitement
antithrombotique, la revascularisation coronaire, et autres traitements
préventifs, le risque d'événements récurrents dans
cette population reste substantiel, en particulier chez les patients
présentant des facteurs de risque plus élevé, comme le
diabète sucré, la dépression du segment ST, ou un score
TIMI
élevé.4
La ranolazine est un dérivé de la pipérazine, qui exerce
des actions antiischémiques sans effet cliniquement significatif sur la
fréquence cardiaque ou la pression
artérielle.5,6
À des concentrations cliniquement adaptées, la ranolazine inhibe
la composante de courant sodique à inactivation lente du myocarde
(I[NaL]), qui pourrait réduire les effets
délétères associés aux surcharges sodique et
calcique intracellulaires qui accompagnent et pourraient promouvoir
l'ischémie
myocardique.7,8
La ranolazine est approuvée dans le traitement antiangineux des
patients souffrant d'angor chronique, mais n'a pas été
étudiée chez les patients avec SCA, ni dans la prévention
secondaire des événements cardiovasculaires majeurs chez les
patients avec maladie coronarienne établie. Une association
établie entre la ranolazine et l'allongement de l'intervalle QT a
suscité des interrogations quant à l'innocuité de la
substance. En conséquence, des données d'innocuité
complémentaires sont nécessaires pour guider son utilisation
chez les patients avec maladie
coronarienne.7
L'étude MERLIN (Metabolic Efficiency With Ranolazine for Less Ischemia
in Non-ST-Elevation Acute Coronary Syndromes)-TIMI 36 a été
élaborée pour évaluer l'efficacité et
l'innocuité de la ranolazine en tant que nouveau traitement visant
à réduire le décès cardiovasculaire, l'infarctus
du myocarde (IDM) ou l'ischémie récurrente, à court et
long terme, chez des patients à risque modéré à
élevé recevant un traitement de référence pour des
SCA.9
MÉTHODES
Population de patients
Entre le 8 octobre 2004 et le 24 mai 2006, 6 560 patients
(Figure 1) ont
été randomisés dans 442 centres de 17 pays (liste
disponible en ligne sur
http://www.jama.com).
Les détails du schéma de l'étude ont été
publiés
précédemment.9
Les patients éligibles étaient âgés de 18 ans ou
plus; ils présentaient des symptômes correspondant à une
ischémie myocardique au repos, pendant au moins 10 minutes et dans les
précédentes 48 heures; et avaient au moins 1 des indicateurs
suivants de risque modéré à élevé de
décès ou d'événements ischémiques
récurrents: élévation des marqueurs biologiques de
nécrose, dépression du segment ST d'au moins 0,1 mV,
diabète sucré, ou score TIMI intermédiaire ou
élevé (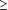 3) pour l'angor instable/IDM sans
élévation du segment
ST.9 Les
patients étaient inéligibles s'ils présentaient l'un des
principaux critères d'exclusion suivants: choc cardiogénique,
élévation persistante du segment ST, revascularisation
réussie de la sténose responsable avant la randomisation,
maladie hépatique cliniquement significative, insuffisance
rénale terminale nécessitant une dialyse, traitement par des
médicaments connus pour prolonger l'intervalle QT, anomalies de
l'électrocardiogramme susceptibles d'interférer avec
l'interprétation de l'enregistrement Holter pour l'ischémie, ou
une espérance de vie de moins de 12
mois.9 La
race et l'ethnicité étaient auto-déclarées, selon
des catégories définies par les investigateurs. Le protocole a
été approuvé par les comités d'éthiques
concernés de tous les centres participants. Le consentement
éclairé écrit a été obtenu auprès de
tous les participants. 3) pour l'angor instable/IDM sans
élévation du segment
ST.9 Les
patients étaient inéligibles s'ils présentaient l'un des
principaux critères d'exclusion suivants: choc cardiogénique,
élévation persistante du segment ST, revascularisation
réussie de la sténose responsable avant la randomisation,
maladie hépatique cliniquement significative, insuffisance
rénale terminale nécessitant une dialyse, traitement par des
médicaments connus pour prolonger l'intervalle QT, anomalies de
l'électrocardiogramme susceptibles d'interférer avec
l'interprétation de l'enregistrement Holter pour l'ischémie, ou
une espérance de vie de moins de 12
mois.9 La
race et l'ethnicité étaient auto-déclarées, selon
des catégories définies par les investigateurs. Le protocole a
été approuvé par les comités d'éthiques
concernés de tous les centres participants. Le consentement
éclairé écrit a été obtenu auprès de
tous les participants.
|
|
|
|
Figure 1.. Organigramme des patients
|
|
|
Protocole de l'étude
Le protocole spécifiait que les patients devaient recevoir un
traitement standard pour les SCA sans surélévation du segment ST
et des mesures de prévention secondaire. Les patients éligibles
étaient randomisés au rapport de 1:1 pour recevoir de la
ranolazine ou un placebo, au moyen d'un système informatisé
central utilisant une méthode de blocs de permutation, avec une
stratification en fonction de la stratégie de traitement initiale
prévue par le médecin responsable (invasive précoce vs
conservatrice), déclarée au moment de la randomisation.
Le traitement étudié devait être administré sous
la forme de 200 mg de ranolazine (ou placebo correspondant) par voie
intraveineuse pendant 1 heure, suivi d'une perfusion de 80 mg/h,
réduite à 40 mg/h pour les patients avec une clairance de la
créatinine inférieure à 30 mL/min (< 0,50 mL/s),
poursuivie pendant 12 à 96 heures. Après administration de la
perfusion, la prise du médicament étudié (ranolazine
à libération prolongée ou placebo correspondant) devait
être poursuivie par voie orale à la dose de 1 000 mg deux fois
par jour, jusqu'à la fin de l'étude. Le protocole autorisait une
réduction posologique pour les patients présentant une
insuffisance rénale, et pour ceux présentant des
événements indésirables spécifiques, susceptibles
d'être liés au traitement, incluant l'allongement persistant de
l'intervalle
QT.9 Pour les
patients avec des ajustements posologiques effectués pendant la
perfusion intraveineuse, le médicament étudié
était poursuivi par voie orale à la dose de 750 mg deux fois par
jour, 500 mg deux fois par jour, ou 375 mg deux fois par jour, en fonction du
débit de perfusion
final9. Un
ajustement posologique supplémentaire pouvait être
effectué en fonction de la persistance ou de la résolution de la
raison de la modification. Les patients revenaient pour les visites de suivi
à 14 jours, 4 mois, puis tous les 4 mois, jusqu'à la fin de
l'étude. Le dernier jour de suivi était le 14 février
2007. Pendant les visites de suivi, les patients étaient
examinés, évalués pour les événements
indésirables et la qualité de vie, et des
prélèvements sanguins étaient effectués pour des
analyses biologiques locales, centrales, ou les deux. Les patients
arrêtant définitivement le médicament étudié
prématurément pendant l'étude étaient suivis par
téléphone. Pour détecter l'ischémie, les patients
ont été équipés, au moment de la randomisation,
d'un enregistreur électrocardiographique numérique de type
Holter (Lifecard CF, Delmar Reynolds, Irvine, Californie), qu'ils devaient
porter pendant 7 jours, période post-hospitalisation incluse. Des
épreuves d'effort étaient effectuées à 8 mois, ou
à la visite finale si elle était antérieure à
cette date, chez les patients aptes à faire de l'exercice.
L'étude devait être poursuivie jusqu'à ce qu'un minimum
de 310 décès et 730 événements cardiovasculaires
majeurs aient été rapportés au centre coordinateur,
délai au-delà duquel il était demandé à
tous les patients de revenir pour une visite de suivi finale.
Critères d'évaluation
Le critère d'efficacité primaire de l'étude
était la première apparition de tout élément du
critère composite décès cardiovasculaire, IDM ou
ischémie récurrente. Le principal critère secondaire
était la première apparition d'un événement
cardiovasculaire majeur, défini par le composite de décès
cardiovasculaire, IDM ou ischémie sévère
récurrente.
L'infarctus du myocarde devait être distinct de
l'événement index, et était défini par des
symptômes suggestifs d'ischémie/infarctus, associés
à des signes électrocardiographiques, des biomarqueurs
cardiaques ou des signes pathologiques d'infarctus, selon des critères
adaptés de la définition élaborée par l'American
College of
Cardiology.9,10
L'ischémie récurrente incluait l'un des critères
suivants: (1) ischémie récurrente avec modifications de
l'électrocardiogramme; (2) ischémie récurrente
entraînant l'hospitalisation; (3) ischémie récurrente
nécessitant une revascularisation, et (4) aggravation de
l'angor/ischémie d'au moins 1 classe d'angor CCS (Canadian
Cardiovascular Society), ayant entraîné l'intensification du
traitement
antiangineux.9
L'ischémie récurrente était considérée
comme sévère si l'un des 3 premiers critères était
rempli. Les autres critères secondaires incluaient l'échec
thérapeutique, défini par le critère composite incluant
le décès cardiovasculaire, l'IDM, l'ischémie
récurrente, le Holter positif pour l'ischémie, l'hospitalisation
pour survenue ou aggravation d'une insuffisance cardiaque, ou une
épreuve d'effort précocement positive (signe d'ischémie
avant d'avoir effectué 12 minutes d'un protocole de Bruce
modifié ou équivalent). La qualité de vie était
évaluée en critère secondaire, à l'aide des
échelles évaluant la fréquence de l'angor et le handicap
physique du questionnaire SAQ (Seattle Angina Questionnaire),11 à 4
mois de suivi. Le critère d'efficacité prédéfini
pour l'évaluation de la phase aiguë jusqu'à 30 jours
était le critère composite de décès
cardiovasculaire, IDM, ischémie sévère récurrente,
ou Holter positif pour l'ischémie.
Les critères d'innocuité incluaient le décès
toutes causes confondues, le critère composite décès
toutes causes confondues ou toute hospitalisation cardiovasculaire,
l'incidence d'arythmie symptomatique documentée, et les arythmies
cliniquement significatives détectées à l'enregistrement
Holter du protocole. Les arythmies symptomatiques documentées
incluaient toute arythmie symptomatique ayant entraîné ou
prolongé une hospitalisation, ou considérée comme
médicalement importante par l'investigateur et documentée par
toute forme d'enregistrement ECG.
Tous les éléments du critère composite primaire et des
principaux critères d'efficacité secondaires, ainsi que
l'hospitalisation pour apparition ou aggravation d'une insuffisance cardiaque,
et les arythmies symptomatiques documentées, ont été
jugés en aveugle par un comité des événements
cliniques.9
Analyses statistiques
L'analyse d'efficacité a consisté en une analyse
hiérarchique des hypothèses primaire puis secondaire dans un
ordre prédéfini, au moyen d'une procédure de tests
fermés. Après l'obtention d'un résultat de test non
significatif, les analyses des critères secondaires restants
étaient considérées comme exploratoires. Ce processus
était destiné à préserver l'erreur de type I
globale définie pour l'ensemble des tests fermés. L'étude
était conçue pour avoir une puissance statistique d'au moins 90
% pour détecter une réduction du risque relatif de 20 % du
principal critère secondaire, sous traitement par ranolazine, en
supposant une incidence de 18 % à 1 an dans le groupe placebo.
Toutes les analyses d'efficacité ont été
effectuées selon le principe de l'intention de traiter. L'analyse du
critère primaire incluait tous les événements primaires
d'efficacité, connus pour être survenus entre la randomisation et
la visite de suivi finale du patient. Les analyses d'efficacité
primaire et secondaire majeure ont été effectuées
à l'aide du test du log-rank, avec une stratification en fonction de
l'intention d'utiliser une stratégie invasive précoce. Les
rapports des risques instantanés (HR) et les intervalles de confiance
(IC) à 95 % ont été estimés par un modèle
de Cox, avec des effets pour le traitement et l'intention d'utiliser une
stratégie invasive précoce. Les taux d'événements
sont présentés en taux d'échec selon Kaplan-Meier
à 12 mois.
Toutes les analyses d'innocuité ont été
effectuées selon le traitement réellement reçu (une seule
dose ou plus) par le patient. Les évaluations périodiques
d'innocuité ont été effectuées par un
comité indépendant de surveillance. Une analyse
d'efficacité intermédiaire programmée, basée sur
le décès cardiovasculaire, a été effectuée
en utilisant une limite d'arrêt de
Fleming-Harrington-O'Brien.12
La valeur de p (test bilatéral) significative était de 0,0497
pour l'analyse d'efficacité primaire, après correction pour
l'analyse intermédiaire.
Notre étude était une étude clinique initiée
par l'investigateur, menée par le groupe d'étude TIMI,
conçue en association avec le comité scientifique, et revue par
le promoteur. Les investigateurs avaient un accès libre et complet aux
données. La coordination des données a été
effectuée par le Nottingham Clinical Research Group (liste disponible
en ligne sur
http://jama.com).
La base de données source a été fournie au groupe
d'étude TIMI et toutes les analyses rapportées dans cet article
ont été effectuées indépendamment par le groupe
d'étude TIMI (S.A.M.), dont les membres ont écrit cet article et
assument la responsabilité des données. La validation des
principales analyses d'efficacité et d'innocuité a
également été effectuée par le Nottingham Clinical
Research Group (centre de coordination des données), ainsi que par le
promoteur. Les analyses ont été menées par le groupe
d'étude TIMI, à l'aide du logiciel Stata SE, version 9.0
(StataCorp LP, College Station, Texas).
RÉSULTATS
Les 2 groupes de patients étaient bien appariés en termes de
caractéristiques initiales (Tableau
1). Au total, 6 303 patients (96,1 %) ont été
traités par aspirine, 5 926 patients (90,3 %) par de l'héparine
non fractionnée ou par une héparine de bas poids
moléculaire, et 955 patients (14,6 %) par un antagoniste des
récepteurs glycoprotéiques IIb/IIIa. Le délai
médian entre l'apparition des symptômes et la randomisation
était de 24 heures (étendue interquartile, 13-34). Le
médicament étudié a été administré
par voie intraveineuse à 6 541 patients (99,7 %) pendant une
médiane de 23 heures (étendue interquartile, 19-29), puis par
voie orale chez 6 399 patients (97,5 %). Les SCA qualifiants ont
été pris en charge par un traitement médical seul chez 3
966 patients (60,5 %), par une intervention coronaire percutanée chez 2
074 patients (31,6 %), et par un pontage aorto-coronarien chez 520 patients
(7,9 %). Pendant la période de traitement, les médicaments
concomitants ont été administrés aux patients comme suit:
clopidogrel ou ticlodipine pour 4 215 patients (64,3 %);?-bloquants pour 5 852
patients (89,2 %); inhibiteurs calciques pour 1 977 patients (30,1 %),
incluant du diltiazem pour 312 patients (4,8 %) et du verapamil pour 190
patients (2,9 %); inhibiteurs de l'enzyme de conversion ou antagonistes de
l'angiotensine II pour 5 130 patients (78,2 %); et statines pour 5 404
patients (82,4 %). Les patients ont été suivis jusqu'à 24
mois, avec un suivi médian de 348 jours (étendue interquartile,
236-460). Neuf patients (0,1 %) ont été perdus de vue.
|
|
|
|
Tableau 1.. Caractéristiques initiales des patients*
|
|
|
Critères d'efficacité
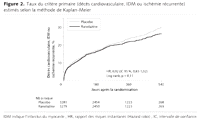
Le critère primaire (décès cardiovasculaire, IDM ou
ischémie récurrente) est survenu chez 696 patients (21,8 %) dans
le groupe ranolazine comparé à 753 patients (23,5 %) dans le
groupe placebo (HR, 0,92; IC 95 %, 0,83-1,02; p = 0,11)
(Figure 2). Le principal
critère secondaire (décès cardiovasculaire, IDM ou
ischémie sévère récurrente) est survenu chez 602
patients (18,7 %) du groupe ranolazine (19,2 %) comparé à 625
patients (19,2 %) du groupe placebo (HR, 0,96; IC 95 %, 0,86-1,08; p = 0,50).
L'échec thérapeutique (décès cardiovasculaire,
IDM, ischémie récurrente, Holter positif pour l'ischémie,
hospitalisation pour apparition ou aggravation d'une insuffisance cardiaque,
ou épreuve d'effort précocement positive a été
observé chez 1 173 patients (36,8 %) dans le groupe ranolazine
comparé à 1 233 patients (38,3 %) dans le groupe placebo (HR,
0,94; IC 95 %, 0,87-1,02; p = 0,16).
|
|
|
|
Figure 2.. Taux du critère primaire (décès cardiovasculaire, IDM
ou ischémie récurrente) estimés selon la méthode
de Kaplan-Meier
|
|
|
Les éléments individuels du critère
d'évaluation primaire et du critère d'échec
thérapeutique, à 30 jours et à la fin de l'étude,
sont présentés dans le
Tableau 2. La ranolazine
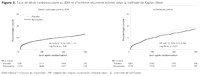
n'avait pas d'effet sur le taux de décès cardiovasculaire ou
d'IDM, pris individuellement ou dans le critère composite
(Figure 3). Cependant,
l'incidence cumulée d'ischémie récurrente était
significativement inférieure chez les patients assignés à
la ranolazine comparé à ceux assignés au placebo
(Figure 3). Une tendance en
faveur d'une réduction précoce des complications
ischémiques récurrentes a été observée avec
la ranolazine sur le critère à 30 jours de décès
cardiovasculaire, IDM, ischémie sévère récurrente,
ou Holter positif pour l'ischémie (p = 0,055)
(Tableau 2). Un effet sur
l'angor du traitement à long terme par ranolazine a été
observé avec plusieurs critères exploratoires
prédéfinis. L'aggravation de l'angor d'au moins 1 classe CCS
(Canadian Cardiovascular Society), nécessitant l'intensification du
traitement médical, était réduite sous ranolazine
comparé au placebo (135 [4,2 %] vs 175 [5,9 %]; HR, 0,77; IC 95 %,
0,62-0,97; p = 0,02). En outre, l'intensification ou l'adjonction d'un
traitement antiangineux était moins fréquente dans le groupe
ranolazine (316 [10,6 %]) comparé au groupe placebo (391 [13,0 %]; HR,
0,80; IC 95 %, 0,69-0,93; p = 0,003). Une faible amélioration dans la
fréquence d'angor sous ranolazine a été observée
avec le questionnaire SAQ (moyenne [ET], 84,3 [22,2] dans le groupe ranolazine
vs 82,2 [23,2] dans le groupe placebo; p < 0,001). Cette différence
était majorée chez les patients inclus dans l'étude avec
une histoire d'angor (n = 2 898 avec les données du SAQ; moyenne (ET]
79,5 [24,1] dans le groupe ranolazine vs 75,5 [25,3] dans le groupe placebo; p
< 0,001). Aucune différence intergroupe n'a été
observée dans l'échelle de handicap physique, sur la population
totale (p = 0,91) comme pour les patients avec antécédent
d'angor (p = 0,30).
|
|
|
|
Tableau 2.. Critères d'efficacité*
|
|
|
|
|
|
|
Figure 3.. Taux de décès cardiovasculaire ou IDM et d'ischémie
récurrente estimés selon la méthode de Kaplan-Meier
|
|
|
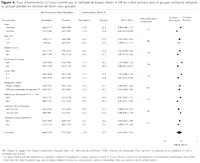
Il n'était pas observé
d'hétérogénéité significative dans l'effet
de la ranolazine sur le critère primaire entre les principaux
sous-groupes analysés, incluant les patients traités par
stratégie non invasive (Figure
4). À la différence des précédentes
études suggérant une efficacité réduite de la
ranolazine sur la performance à l'effort chez les femmes avec angor
stable,13 l'effet sur le critère primaire était significatif
chez les femmes (n = 2 291; HR, 0,83; IC 95 %, 0,70-0,99), avec une
réduction relative de 29 % de l'ischémie récurrente avec
la ranolazine (p = 0,002), mais sans donnée statistique
déterminante d'une interaction basée sur le sexe (p = 0,12 pour
l'interaction). Aucune hétérogé-néité dans
l'effet de la ranolazine sur l'ischémie récurrente n'a
été observée chez les patients traités par une
stratégie invasive précoce comparé à une
stratégie conservatrice (p = 0,52 pour l'interaction).
|
|
|
|
Figure 4.. Taux d'événements (12 mois) estimés par la
méthode de Kaplan-Meier et HR du critère primaire dans le groupe
ranolazine comparé au groupe placebo en fonction de divers
sous-groupes
|
|
|
Innocuité et tolérance
Dans la population de l'analyse d'innocuité, le décès
toutes causes confondues ne différait pas entre les patients
traités par ranolazine et ceux traités par placebo (HR, 0,99; IC
95 %, 0,80-1,22; p = 0,91) (Tableau
3). La mort subite cardiaque et le critère composite
décès toutes causes confondues ou toute hospitalisation
cardiovasculaire ne différaient pas non plus entre les patients
traités par ranolazine (56 [1,7 %] et 1 046 [33,2 %] respectivement) et
ceux traités par placebo (respectivement 65 [1,8 %] et 1 082 [33,4 %]).
L'incidence d'arythmies symptomatiques documentées tout au long de
l'étude était similaire chez les patients du groupe ranolazine
et ceux du groupe placebo (p = 0,84). En outre, la fréquence des
arythmies cliniquement significatives observée dans l'enregistrement
Holter (n = 6 351) des 7 premiers jours était inférieure dans le
groupe ranolazine (2 330 patients [73,7 %]) vs groupe placebo (2 650 patients
[83,1 %], p < 0,001). Cette réduction incluait une incidence
inférieure de tachycardie ventriculaire (respectivement 948 [30 %] sur
3 158 patients vs 1 211 [38 %] sur 3 184 patients; p < 0,001).
|
|
|
|
Tableau 3.. Principaux critères d'innocuité*
|
|
|
L'arrêt du traitement motivé par un événement
indésirable, le choix du patient, ou pour toute autre raison, est
survenu chez 915 patients (28 %) du groupe ranolazine et 736 patients (22 %)
du groupe placebo (p < 0,001). L'arrêt dû à un
événement indésirable a été rapporté
significativement plus souvent chez les patients sous ranolazine (286 [8,8 %])
que chez ceux traités par placebo (154 [4,7], p < 0,001). Pendant le
traitement, la réduction permanente de la dose du médicament
étudié pendant la phase intraveineuse, pour cause
d'événement indésirable, a été
observée chez 63 patients (1,9 %) traités par ranolazine et 37
patients (1,1 %) sous placebo. En outre, chez 13 patients (0,4 %) du groupe
ranolazine, une réduction posologique a été
effectuée pour dysfonction rénale; chez 31 patients (0,9 %),
elle a été effectuée pour cause d'allongement persistant
de l'intervalle QTc, et chez 11 patients (0,3 %), pour d'autres raisons. Dans
le groupe placebo, chacune de ces proportions était respectivement de 9
(0,3 %), 10 (0,3 %) et 11 (0,3 %) patients. Au cours du traitement chronique
par voie orale avec le médicament étudié, la dose a
été réduite chez 334 patients (10 %) recevant la
ranolazine et chez 177 patients (5 %) recevant le placebo (p < 0,001). Dans
le groupe ranolazine, la dernière dose prise était de 1 000 mg
deux fois par jour chez 2 715 patients (83 %), 750 mg deux fois par jour chez
180 patients (6 %), 500 mg deux fois par jour chez 235 patients (7 %), et 375
mg deux fois par jour chez 64 patients (2 %); 74 patients (2 %) n'ont jamais
pris de dose orale.
Les événements indésirables les plus fréquents,
qui n'étaient pas des critères d'évaluation, survenus
chez plus de 4 % des patients, et plus fréquents sous ranolazine que
sous placebo, étaient les vertiges (13 % vs 7 %), les nausées (9
% vs 6 %), et la constipation (9 % vs 3 %). Cent neuf cas de syncope ont
été observés dans le groupe ranolazine (3,3 %), et 75 cas
dans le groupe placebo (2,3 %; p = 0,01). Ces cas incluaient les
événements rapportés comme syncope, syncope vaso-vagale,
et perte de conscience. Le nombre supérieur de cas de syncope dans le
groupe ranolazine (n = 34) était majoritairement classifié par
l'investigateur en syncope vaso-vagale (respectivement 38 cas vs 18 cas). Deux
cas de torsades de pointe ont été identifiés par les
investigateurs, dont 1 dans le groupe placebo et 1 dans le groupe
ranolazine.
COMMENTAIRES
Dans cette étude de patients avec SCA sans
surélévation du segment ST, à risque modéré
à élevé d'événements cardiovasculaires
récurrents, aucun bénéfice significatif de la ranolazine
n'a été observé comparé au placebo, en ce qui
concerne le critère composite de décès cardiovasculaire,
IDM ou ischémie récurrente, pendant une médiane d'un an
de traitement. Les analyses complémentaires ont
révélé une réduction relative de 13 % du risque
d'ischémie récurrente, et une moindre intensification des autres
traitements antiangineux chez les patients traités par ranolazine,
parallèlement à une absence d'effet sur le composite de
décès cardiovasculaire ou IDM. La ranolazine s'est
révélée sans danger, et aucune différence n'a
été observée dans les critères d'innocuité
prédéfinis d'arythmie symptomatique documentée, de mort
subite cardiaque, ou de décès toutes causes confondues,
comparé au placebo. En effet, la ranolazine était
associée à une réduction significative de la
fréquence d'arythmies détectées par l'enregistrement
Holter des 7 premiers jours suivant la randomisation. Davantage de cas de
syncope ont été rapportés avec la ranolazine, en accord
avec la précédente étude effectuée chez des
patients avec angor
chronique.7
La ranolazine est actuellement approuvée dans le traitement de
patients sélectionnés, présentant un angor chronique et
des symptômes persistant malgré un traitement par
β-bloquants, inhibiteurs calciques ou nitrates. De
précédentes études de la ranolazine à
libération prolongée ont été menées chez
des patients atteints de maladie coronarienne confirmée,
présentant une dépression du segment ST avant l'accomplissement
de 9 minutes d'un protocole de Bruce
modifié,5,6
ou au moins 3 épisodes d'angor par semaine malgré un traitement
par inhibiteur
calcique.14
Ces études ont conjointement démontré que la ranolazine
prolonge le délai jusqu'à l'ischémie à
l'épreuve d'effort sur tapis roulant, améliore la durée
de l'effort, et réduit la fréquence d'angor ainsi que
l'utilisation de nitroglycérine sublinguale chez ces patients hautement
symptomatiques.7
Les analyses en sous-groupes de ce études (environ 1 500 patients au
total) ont indiqué une possible réduction de l'effet traitement
de la ranolazine sur la performance à l'effort chez les
femmes.13 En
raison d'une augmentation dose-dépendante de l'intervalle QT,
l'utilisation de la ranolazine a été uniquement
recommandée chez les patients n'ayant pas obtenu de réponse
adéquate à d'autres
antiangineux.7
En outre, dans ces études, la taille de l'échantillon et la
durée du traitement n'étaient pas conçues pour
évaluer l'effet de la ranolazine dans la prévention secondaire
des événements cardiovasculaires majeurs. Des données
expérimentales ont révélé des réductions
dans l'étendue des lésions ischémiques et une
amélioration de la performance ventriculaire gauche dans des
modèles animaux avec IDM
aigu.15
Cependant, la ranolazine n'a pas été précédemment
étudiée chez des patients avec syndromes ischémiques
aigus.
Nous avons recruté des patients à l'admission pendant la
phase aiguë de leur SCA, et avons étudié
l'efficacité de la ranolazine pour une nouvelle application potentielle
dans la prise en charge aiguë des SCA, ainsi que dans la
prévention à long terme des événements
cardiovasculaires majeurs et de l'ischémie récurrente. Les
résultats de cette étude randomisée de forte puissance ne
soutiennent pas l'utilisation de la ranolazine dans la prise en charge
aiguë des SCA ou comme un traitement modifiant l'évolution de la
maladie pour la prévention secondaire du décès
cardiovasculaire ou de l'IDM. Cependant, nos résultats suggèrent
un bénéfice de la ranolazine comme traitement antiangineux dans
une population de patients substantiellement plus vaste que les
précédentes études, avec une cardiopathie
ischémique établie. Les analyses en sous-groupes doivent
être interprétées avec une certaine réserve, compte
tenu du résultat d'efficacité primaire globalement non
significatif. Néanmoins, à la différence des
précédentes études basées sur les épreuves
d'effort, la réduction de l'ischémie récurrente avec la
ranolazine n'était certainement pas inférieure chez les femmes
comparées aux hommes.
Dans cette grande étude, qui double quasiment les données
d'innocuité existant sur la ranolazine à libération
prolongée, nous avons trouvé une absence de majoration des
arythmies ou des morts subites cardiaques chez les patients traités par
ranolazine comparé au placebo, pendant un suivi médian d'un an.
Un taux supérieur d'arrêt dû à des
événements indésirables a été
observé dans le groupe ranolazine, les événements
indésirables les plus fréquents étant les vertiges, les
nausées, et la constipation. Ce profil de tolérance,
parallèlement à la proportion majorée de patients avec
syncope, doivent être pris en compte par le clinicien dans
l'évaluation des risques et des bénéfices potentiels du
traitement par ranolazine. L'étiologie de la syncope sous ranolazine
reste inexpliquée et nécessite des études
complémentaires.7
L'observation d'une réduction significative des arythmies,
détectées sur enregistrement Holter des 7 premiers jours,
apporte la première preuve clinique de la pertinence potentielle de
données expérimentales montrant la suppression de marqueurs de
proarythmie, incluant la post-dépolarisation précoce et la
dispersion transmurale de la repolarisation, avec la
ranolazine;16
elle permet en outre de modérer, dans une certaine mesure, les craintes
quant à la responsabilité potentielle de l'arythmie dans la
syncope. Néanmoins, la sensibilisation à son utilisation
à plus long terme dans la communauté reste importante. Les
effets anti-arythmiques potentiels de la ranolazine justifient des
études complémentaires.
Les limites suivantes de notre étude doivent être reconnues.
Toutes les analyses d'efficacité rapportées dans cet article
étaient prédéfinies dans le cadre du plan d'analyse
statistique qui a été finalisé avant le verrouillage de
la base de données. Compte tenu du résultat statistiquement non
significatif pour le critère d'évaluation primaire, toutes les
analyses d'efficacité complémentaires, bien que
prédéfinies, doivent être considérées comme
exploratoires de facto. Cependant, particulièrement lorsqu'elles sont
interprétées dans le contexte des précédentes
études randomisées sur la
ranolazine,5,6,14
nos observations relatives à l'efficacité et à
l'apparente innocuité de la ranolazine en tant qu'antiangineux
contribuent à une définition de son utilisation clinique. Dans
notre étude, la fréquence de l'arrêt
prématuré définitif du médicament
étudié était comparable à celle d'autres
études contemporaines du traitement à long terme après
présentation avec
SCA.17,18
On pourrait penser que l'arrêt prématuré du
médicament étudié a pu biaiser l'analyse
d'efficacité en intention de traiter en faveur du résultat
nul.
CONCLUSIONS
L'adjonction de la ranolazine au traitement de référence
actuel des SCA sans surélévation du segment ST n'a pas
démontré d'efficacité dans la réduction du taux du
critère composite associant le décès cardiovasculaire,
l'IDM et l'ischémie récurrente, et n'est pas indiqué dans
le traitement des SCA. La réduction de l'ischémie
récurrente, observée dans une large population de patients avec
maladie coronarienne établie, s'accorde avec de
Précédentes données chez des patients
sélectionnés avec angor chronique. Ces résultats,
associés au profil d'innocuité global favorable observé,
fournissent de nouvelles données permettant de guider l'utilisation de
la ranolazine dans le traitement antiangineux des patients souffrant d'angor
chronique.
Informations sur les auteurs
Correspondance: David A. Morrow, MD, MPH, TIMI Study Group, Department
of Medicine, Brigham and Women's Hospital, 75 Francis St, Boston, MA 02115
(dmorrow{at}partners.org).
Contributions des auteurs: Les Drs Morrow et Braunwald et Ms Murphy
ont eu un accès complet à toutes les données de
l'étude et acceptent la responsabilité de
l'intégrité des données et de l'exactitude de l'analyse
des données.
Conception et schéma de l'étude: Morrow,
Karwatowska-Prokopczuk, Wolff, Skene, McCabe, Braunwald.
Recueil des données data: Morrow, Scirica, Skene, McCabe,
Braunwald.
Analyse et interpretation des données: Morrow, Scirica,
Karwatowska-Prokopczuk, Murphy, Budaj, Varshavsky, Wolff, Skene, McCabe,
Braunwald.
Rédaction du manuscrit: Morrow, Braunwald.
Revue critique du manuscrit: Morrow, Scirica,
Karwatowska-Prokopczuk, Murphy, Budaj, Varshavsky, Wolff, Skene, McCabe,
Braunwald.
Analyse statistique: Murphy, Skene.
Recueil du financement: Morrow, McCabe, Braunwald. Aide
administrative, technique ou matérielle: Morrow, Scirica,
Karwatowska-Prokopczuk, Murphy, Skene, McCabe, Braunwald.
Supervision de l'étude: Morrow, McCabe, Braunwald.
Liens financiers: Le groupe d'étude TIMI déclare avoir
reçu une bourse de recherche de Accumetrics, Amgen, AstraZeneca, Bayer
Healthcare, Beckman Coulter, Biosite, Bristol-Myers Squibb, CV Therapeutics,
Eli Lilly, GlaxoSmithKline, Inotek Pharmaceuticals, Integrated Therapeutics,
Merck & Co, Merck-Schering Plough Joint Venture, Millennium
Pharmaceuticals, Novartis Pharmaceuticals, Nuvelo, Ortho-Clinical Diagnostics,
Pfizer, Roche Diagnostics, Sanofi-Aventis, Sanofi-Synthelabo, et
Schering-Plough. Le Dr Morrow déclare avoir reçu des honoraires
pour des présentations de formation médicale continue de CV
Therapeutics et Sanofi-Aventis, d'avoir été consultant pour
GlaxoSmithKline et Sanofi-Aventis, et de siéger au comité
consultatif de Genentech. Le Dr Scirica déclare avoir reçu des
honoraires pour des présentations de formation médicale de CV
Therapeutics. Le Dr Karwatowska-Prokopczuk est sa propre employée et
possède des actions chez CV Therapeutics. Le Dr Budaj déclare
avoir reçu des honoraires de AstraZeneca, CV Therapeutics,
GlaxoSmithKline, et Sanofi-Aventis, et être consultant chez
GlaxoSmithKline et Sanofi-Aventis. Le Dr Varshavsky déclare avoir
reçu des bourses de recherche de CV Therapeutics. Le Dr Wolff
était salarié chez CV Therapeutics et actuellement travaille
comme consultant et détient des parts chez CV Therapeutics, il
déclare avoir son nom sur des licences pour la ranolazine. Le Dr
Braunwald déclare avoir reçu des honoraires de et d'être
consultant chez AstraZeneca, Bayer AG, Daichii Sankyo, Merck, Pfizer, et
Schering-Plough. Les Dr Skene et Mss Murphy et McCabe n'ont rapporté
aucun lien financier.
Financement/Soutien: L'essai MERLIN-TIMI 36 a
bénéficié du soutien de CV Therapeutics.
Rôle du sponsor: Le protocole était développé
par le groupe d'étude TIMI Study en conjonction avec le comité
scientifique et revu par le sponsor de l'étude. Les salariés du
sponsor ont travaillé avec les investigateurs pour préparer le
plan d'analyse statistique. Toutes les analyses primaires ont
été réalisées par le groupe d'étude TIMI
avec validation par le Nottingham Clinical Research et le sponsor. Les
salaries du sponsor ont revu le manuscrit ensemble avec les co-auteurs et ont
fait des suggestions d'édition sans obligation de suivi.
Analyse statistique indépendante: Les investigateurs ont eu
un accès libre et complet aux données. La coordination des
données a été réalisée par le Groupe
Nottingham Clinical Research (voir annexe en ligne). La base de données
brutes a été fournie au groupe d'étude TIMI et toutes les
analyses rapportées dans ce manuscrit ont été
réalisées de façon indépendante par le groupe
d'étude TIMI (Ms Murphy), dont les membres ont écrit l'article
et acceptent la responsabilité des données. La validation des
analyses de l'efficacité principale et des analyses de tolérance
a été aussi réalisée par Nottingham Clinical
Research (centre de coordination des données), de même que par le
sponsor.
Les investigateurs de l'étude MERLIN-TIMI 36 sont
indiqués en ligne à
http://www.jama.com.
Affiliations des auteurs: TIMI Study Group, Department of
Medicine, Brigham and Women's Hospital, and Harvard Medical School, Boston,
Mass; CV Therapeutics, Palo Alto, Calif; Postgraduate Medical School,
Department of Cardiology, Grochowski Hospital, Warsaw, Poland; Evidence
Clinical and Pharmaceutical Research, St Petersburg, Russia; Cytokinetics, San
Francisco, Calif; Nottingham Clinical Research Limited, Nottingham, United
Kingdom.
BIBLIOGRAPHIE
| |
1. Braunwald E. Unstable angina: an etiologic approach to management.
Circulation.1998; 98:2219
-2222.
FREE FULL TEXT
2. Braunwald E, Antman EM, Beasley JW, et al. ACC/AHA 2002 guideline
update for the management of patients with unstable angina and non-ST-segment
elevation myocardial infarction—summary article: a report of the
American College of Cardiology/American Heart Association Task Force on
practice guidelines (Committee on the Management of Patients With Unstable
Angina). J Am Coll Cardiol. 2002;40
: 1366-1374.
FREE FULL TEXT
3. Gibbons RJ, Abrams J, Chatterjee K, et al. ACC/AHA 2002 guideline
update for the management of patients with chronic stable angina—summary
article: a report of the American College of Cardiology/American Heart
Association Task Force on practice guidelines (Committee on the Management of
Patients With Chronic Stable Angina). J Am Coll
Cardiol. 2003;41:159
-168.
FREE FULL TEXT
4. Antman EM, Cohen M, Bernink PJ, et al. The TIMI risk score for
unstable angina/non-ST elevation MI: a method for prognostication and
therapeutic decision making. JAMA.2000; 284:835
-842.
FREE FULL TEXT
5. Chaitman BR, Pepine CJ, Parker JO, et al. Effects of ranolazine
with atenolol, amlodipine, or diltiazem on exercise tolerance and angina
frequency in patients with severe chronic angina: a randomized controlled
trial. JAMA.2004; 291:309
-316.
FREE FULL TEXT
6. Chaitman BR, Skettino SL, Parker JO, et al. Anti-ischemic effects
and long-term survival during ranolazine monotherapy in patients with chronic
severe angina. J Am Coll Cardiol.2004; 43:1375
-1382.
FREE FULL TEXT
7. Chaitman BR. Ranolazine for the treatment of chronic angina and
potential use in other cardiovascular conditions.
Circulation.2006; 113:2462
-2472.
FREE FULL TEXT
8. Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium
current as a potential cardioprotective principle: effects of the late sodium
current inhibitor ranolazine. Heart.2006; 92(suppl 4):iv6
-iv14.
FREE FULL TEXT
9. Morrow DA, Scirica BM, Karwatowska-Prokopczuk E, Skene A, McCabe
CH, Braunwald E. Evaluation of a novel anti-ischemic agent in acute coronary
syndromes: design and rationale for the Metabolic Efficiency with Ranolazine
for Less Ischemia in Non-ST-elevation acute coronary syndromes (MERLIN)-TIMI
36 trial. Am Heart J.2006; 152:400
-406.
10. Cannon CP, Battler A, Brindis RG, et al. American College of
Cardiology key data elements and definitions for measuring the clinical
management and out-comes of patients with acute coronary syndromes: a report
of the American College of Cardiology Task Force on Clinical Data Standards
(Acute Coronary Syndromes Writing Committee). J Am Coll
Cardiol. 2001; 38:2114
-2130.
FREE FULL TEXT
11. Spertus JA, Jones P, McDonell M, Fan V, Fihn SD. Health status
predicts long-term outcome in out-patients with coronary disease.
Circulation.2002; 106:43
-49.
FREE FULL TEXT
12. Fleming TR, Harrington DP, O'Brien PC. Designs for group sequential
tests. Control Clin Trials.1984; 5:348
-361.
PUBMED
13. Wenger NK, Chaitman B, Vetrovec GW. Gender comparison of efficacy
and safety of ranolazine for chronic angina pectoris in four randomized
clinical trials. Am J Cardiol.2007; 99:11
-18.
PUBMED
14. Stone PH, Gratsiansky NA, Blokhin A, Huang IZ, Meng L. Antianginal
efficacy of ranolazine when added to treatment with amlodipine: the ERICA
(Efficacy of Ranolazine in Chronic Angina) trial. J Am Coll
Cardiol. 2006;48:566
-575.
FREE FULL TEXT
15. Gralinski MR, Black SC, Kilgore KS, Chou AY, McCormack JG, Lucchesi
BR. Cardioprotective effects of ranolazine (RS-43285) in the isolated perfused
rabbit heart. Cardiovasc Res.1994; 28:1231
-1237.
FREE FULL TEXT
16. Antzelevitch C, Belardinelli L, Zygmunt AC, et al.
Electrophysiological effects of ranolazine, a novel antianginal agent with
antiarrhythmic properties. Circulation.2004; 110:904
-910.
FREE FULL TEXT
17. Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate
lipid lowering with statins after acute coronary syndromes. N Engl
J Med. 2004;350:1495
-1504.
PUBMED
18. de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a
delayed conservative simvastatin strategy in patients with acute coronary
syndromes: phase Z of the A to Z trial. JAMA.2004; 292:1307
-1316.
FREE FULL TEXT
ARTICLE EN RAPPORT
La ranolazine a-t-elle une place dans le traitement des syndromes coronaires aigus?
L. Kristin Newby et Eric D. Peterson
JAMA. 2007;297:1823-1825.
Texte Complet
|