Neurological Disease: Listening to Gene Silencers
Abstract
Neurons regulate the expression of genes essential to individual neuron function through elegant combinatorial interactions among a limited number of transcription factors. In addition, an economy of regulatory control is practiced within the nucleus that belies conceptual divisions of transcription factors into “repressors” and “activators.” Studies of the neural restrictive silencer element (NRSE, also known as RE1) and its repressor protein have revealed a multitude of mechanisms by which transcriptional regulation is not only elaborated in normal neuronal development, but perverted in disease states.

Cell differentiation and specialization depend on the concerted expression and repression of specific genes. Investigations
of neuronal gene regulation are unifying mechanisms of up- and downregulation of gene expression that have previously been
regarded as disparate.
INTRODUCTION
“Adjust inhibition!” is a regulatory imperative that can be issued at all organizational levels, from cortical circuitry to transcription. In cortical circuitry, for example, the benzodiazepines elicit their anti-anxiety and anticonvulsant effects by gently nudging GABA (γ-aminobutyric acid) receptors (1). Inhibition of certain NMDA (N-methyl-d-aspartate) receptors by protons is half-maximal at the ambient pH of 7.4, so that slight alterations in extracellular pH can efficiently regulate synaptic excitation (2, 3). Turning to metabolism, the galactose utilization pathway in yeast is a good case in point of activation by disinhibition. The transfer of yeast cultures from glucose- to galactose-rich medium derepresses numerous genes that are under the control of the Mig1 and Gal repressor proteins (4). In many mammalian tissues, the E2F transcription factor (essential for entry into the S-phase of the cell cycle) is normally inhibited by the Rb (retinoblastoma) protein; the inhibitory activity of the Rb protein is removed through its phosphorylation by a cyclin-dependent cyclase (5).
Many gene sequences contain silencers, regulatory elements that, when bound by their cognate repressor, turn down gene expression. REST (RE1 silencing transcription factor), also known as NRSF (neuron-restrictive silencer factor) is a neuronally active repressor that binds to its cognate DNA silencer element, NRSE (or RE1). In this review REST and NRSF will be used interchangeably. Recent and emerging data point to a much broader range of cellular functions for the NRSF–NRSE system than originally envisioned. The question of whether lessons learned from the NRSF–NRSE system can be generalized to other repressor–silencer systems, and whether such systems might become therapeutic drug targets, is a focus of this review. Studies of the neuron-specific regulation of gene expression are essential to an understanding of neuronal phenotype determination and may thus lay the ground for manipulating stem-cell differentiation for the treatment of neurological disorders or for promoting survival following neuronal injury.
REST/NRSF: FUNCTIONS IN THE NERVOUS SYSTEM
Elucidation of the mechanisms that control neuronal gene expression is important for understanding how the brain learns or responds to insults. The NRSE is a 21-bp DNA sequence (Figure 1A⇓) originally isolated from the promoters of the genes that express, primarily in neurons, the type II sodium channel (6, 7) and SCG10 (8, 9). Numerous genes with expression restricted mainly to the nervous system contain an NRSE motif. A Blast search of the Celera mouse database identifies at least 324 genes with NRSE consensus sequences, and functional analysis of cloned promoter sequences reveal that NRSE consensus sequences reside in a diverse set of genes of interest to neuroscience (Table 1⇓).
Selected NRSE-Containing Genes of Interest to Neuroscience
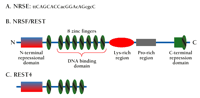
The NRSE–NRSF system.
A. Consensus sequence of the NRSE deduced from functional analysis of silencer elements in nineteen genes (57). Nucleotides in lower case vary frequently among functional silencer elements. B. Domains of the NRSF protein (also known as REST).C. REST4 is a C-terminally truncated splice variant of the NRSF protein.
Although the major role of NRSF was originally thought to be in non-neuronal cells, where it in fact functions as a repressor of neuron-specific genes (7, 9–11), increasing evidence suggests that NRSF may regulate the expression of NRSE-containing genes in neurons as well. First, NRSF mRNA is found in central neurons in specific brain regions both during development and in the adult brain (13–15). Second, mutation or deletion of the NRSE in any one of three distinct neuronal genes failed to elicit widespread ectopic gene expression (15–17). Likewise, although a targeted mutation of the gene encoding NRSF in mice derepressed the expression of neuron-specific tubulin in a subset of non-neural tissues, the ectopic expression of other NRSE-containing genes was not widespread (12). Third, transcription through promoters I and II of the BDNF gene (encoding brain-derived neurotrophic factor) was increased in hippocampal neurons of transgenic mice carrying a mutant NRSE in the BDNF gene (15). Furthermore, induction of BDNF expression from promoters I and II by kainate treatment was remarkably potentiated when the NRSE element was deleted. Together, these findings indicate that a major role of the NRSF–NRSE system may be to regulate gene expression within neurons, in addition to suppressing ectopic expression of neuronal genes outside the nervous system.
NRSF is a member of the GLi-Krüppel family of transcriptional zinc-finger proteins. NRSF contains a cluster of eight zinc-finger repeats near its N terminus, followed by a region rich in basic amino acids, a cluster of six proline-rich repeats, and a single zinc finger near the C terminus (Figure 1B⇑) (18). A number of splice variants of NRSF exist, including REST4 (Figure 1C⇑), a neuron-specific variant lacking the C-terminal repression domain (14). As described above, it is increasingly apparent that NRSF regulates gene expression in neurons, but several important questions remain: a) How does NRSF repress gene expression and is this process itself subject to regulation? b) Which genes are regulated by NRSF in mature differentiated neurons and how is regulation cued? and c) Is NRSF more than just a repressor?
Dual Mechanisms of Repression by NRSF
Deletion and GAL4-fusion analyses have demonstrated two independent repressor domains of NRSF, one at each terminus (Figure 1B⇑) (18, 19). Several independent studies have recently shown that the transcriptional repression mediated by the N terminus of NRSF involves the recruitment of mSin3A/B and a histone deacetylase (HDAC) complex. In yeast, the repression of reporter promoter activity by NRSF requires the presence of SIN3 and RPD3 (mammalian Sin3A/B and HDAC orthologs, respectively) (20–22). Similarly, in mammalian cells, transcriptional repression of NRSE-containing genes in non-neuronal cells is relieved when histone deacetylase activity is inhibited by trichostatin A (22, 23). Tricostatin A treatment also induces endogenous NRSE-containing genes and attenuates the repression activity of exogenous NRSF (22, 23). Furthermore, endogenous NRSF co-immunoprecipitates with endogenous HDAC1, HDAC2, and mSin3A (21, 23, 24), and in vitro, NRSF binds mSin3B (22).
Together, these data support a model in which the N-terminal domain of NRSF recruits HDAC via Sin3B and Sin3A (Figure 2⇓). Modification of histone N-terminal tails by acetylation and deacetylation represents a major pathway for transcription regulation. Poorly acetylated histones are associated with condensed, transcriptionally inactive genes, whereas highly acetylated histones are associated with genes poised for transcription (25, 26). Deacetylation of histones H3 and H4 is thought to induce a condensed chromatin structure that shields promoters from transcription factors and RNA polymerases (27, 28), explaining the action of transcriptional repressors that act by recruiting HDAC complexes.
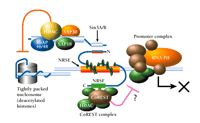
Dual mechanism of repression via the NRSF–NRSE interaction. The N-terminal domain of NRSE-bound NRSF interacts with Sin3, which in turn recruits histone deacetylase (HDAC) and associated proteins (SAP30, SAP18, RbAp46/48) to the promoter region. HDAC deacetylates lysine residues of nucleosomal core histones, which consequently limits the accessibility of DNA to transcription factors; the resulting inhibition of RNA polymerase activity (RNA PII) is indicated schematically by an X. In addition, a zinc-finger domain proximal to the C terminus of NRSF interacts with CoREST. CoREST recruits an HDAC complex that includes HDAC1 and HDAC2 and mediates repression that is distinguished by resistance to trichostatin A. Multiple corepressor mechanisms are thus indicated; see text for details.
These data account for repression by the N-terminal domain of REST, but how about the C-terminal repressor domain? A novel protein, termed CoREST, was recently shown to interact with the C-terminal repressor domain of REST to mediate repression (29). CoREST is a component of a novel HDAC complex (30-32). Trichostatin A fails to prevent the REST C-terminal domain from repressing transcription of both the GluR2 glutamate receptor gene and a gene encoding a GAL4–SCG10 fusion construct (22, 23), but did relieve such repression of the gene that encodes the type II sodium channel (32). The finding that trichostatin A reduces repressor activity of the REST C terminus in some but not all genes suggests that multiple repressor mechanisms may exist for the C terminus of REST.
Regulation of NRSF Function Following Seizures
Evidence is accumulating that NRSF is itself regulated by neuronal activity in the adult brain, consistent with a role for the NRSF–NRSE system in modulating gene expression dynamically rather than simply acting as an on/off switch during development. For example, the expression of REST mRNA is increased in hippocampal and cortical neurons as soon as four hours after kainate-induced seizures begin (Figure 3⇓) (14). Intriguingly, in response to kainate-induced seizures, BDNF transcript levels are also upregulated in the hippocampus within three hours. BDNF is a member of the neurotrophin family that plays roles in neuronal differentiation, survival, and neuronal plasticity. Timmusk et al. (15) investigated the role of the BDNF NRSE using transgenic reporter constructs in a kainate seizure model. The BDNF gene has four promoters that show differential usage in a tissue- and brain region–specific manner (15, 33). Promoters I and II of the BDNF gene, including an intervening NRSE, when fused to a CAT gene (encoding chloramphenicol acetyltransferase as a gene reporter), were sufficient to recapitulate endogenous BDNF expression patterns in transgenic mice. This construct was also sufficient to allow appropriate upregulation in response to kainate. Following disruption of the NRSE, however, rates of induction by kainate were increased fourfold, relative to the wild-type NRSE control construct. This observation suggests that NRSF acts to temper peak BDNF expression levels during seizure, but is not necessary for the induction itself.

Expression of REST mRNA in the rat dentate gyrus (DG) before (left) and after (right) kainate-induced status epilepticus. The expression of truncated variants REST1 and REST4 is also increased in the hippocampus by 4 hours. From (14), with permission.
Another example of a seizure-regulated gene is GluR2, which encodes the AMPA-type (i.e., responsive to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) glutamate receptor subunit that regulates Ca2+ permeability (34). GluR2 expression declines in selected hippocampal neurons within twelve to twenty-four hours after seizure (35). A functional NRSE in the GluR2 promoter has been characterized that inhibits the gene's expression by recruiting HDAC (23, 36), and histones associated with the GluR2 promoter are hypoacetylated in a trichostatin A–sensitive manner within three hours of seizure onset (37). These findings are consistent with the hypothesis that rapidly induced NRSF associates with the GluR2 NRSE to bring about the deacetylation of GluR2-associated histones, and thereby contributes to the observed decline in GluR2 levels with subsequent apoptosis. Testing this hypothesis will require a detailed transgenic study of the GluR2 gene in a seizure model as was performed for the BDNF gene.
By limiting peak and basal expression levels of a number of NRSE-containing neuronal genes (e.g., BDNF and GluR2) NRSF may be a key modulator of neuroprotection. Indeed, NRSF-null mice undergo apoptosis during embryogenesis (12). Identification of the NRSF target genes responsible for cell death may thus suggest neuroprotective strategies for the adult injured brain.
NRSF, REST4, and Cancer
Aberrantly high levels of NRSF have been found in some of the most aggressive tumors (38, 39), and several observations raise the possibility that NRSF is involved in tumor growth. Medulloblastoma is the most malignant pediatric brain tumor, believed to originate from the undifferentiated external layer of the cerebellum. Whether NRSF in some way contributes to this tumorigenic state was investigated by infecting tumors with adenoviral constructs expressing a modified NRSF, the N- and C-terminal repressor domains of which had been replaced with the activation domain of the VP16 transcription factor; the purpose was to derepress the transcription of any genes affected by high NRSF levels. Infection by the adenoviral constructs drastically reduced tumor size and concomitantly activated caspase activity and apoptosis (39). These results suggest that high NRSF levels in some medulloblastomas contribute to the disease state, but the mechanism has yet to be addressed. Given that NRSF regulates many genes that are normally expressed in adult neurons, it is tempting to speculate that NRSF may also target genes involved in terminal differentiation; the expression of such early genes would be consistent with the observed decrease in NRSF levels that occurs in differentiated neurons. Aberrantly high expression of this repressor would thus shut down differentiation genes and so contribute to tumor formation. Identification of NRSF target genes that commit cells to terminal differentiation would allow a test of this hypothesis.
In the context of tumorigenesis and developmental regulation, it is intriguing that the NRSF gene can be alternatively spliced to generate truncated proteins that encompass the N-terminal repression domain and either the first four or five of the eight zinc fingers of the DNA binding domain (REST1 or REST4, respectively). If REST1 and REST4 can bind the NRSE, they do so weakly, as judged by electrophoretic mobility shift assay (14). High levels of REST4 are found in biopsies taken from patients suffering from the highly aggressive Small Cell Lung Carcinoma (SCLC) (38). SCLCs are characterized by the expression of neuropeptides, such as arginine vasopressin (AVP), that can cause hyponatremia syndrome and may contribute to tumor growth via an autocrine loop (40). The AVP gene possesses an NRSE (41), and AVP promoter–reporter gene constructs that contain this NRSE are repressed in non-SCLC tumor cells that express NRSF. However, in SCLC cells that express predominantly more REST4 than NRSF, the same reporter is expressed, suggesting that REST4 can antagonize NRSF-mediated repression. Interestingly, REST4 has been reported to derepress expression of an NRSE-containing reporter construct in neuroblastoma cells (42) as well as a cholinergic gene locus in PC12 cells (43). Certain medullo- and neuroblastomas may thus develop according to a model in which REST4 mediates the derepression of target genes, rather than to a model in which NRSF would predominate as a repressor. Identification of the relevant NRSF-targeted genes will be important for selecting between these alternative hypotheses.
NRSF as a Multifunctional Protein
The observations described above with tumor cells suggest that REST4 may be able to squelch NRSF-mediated repression by sequestering away the Sin3/HDAC complex to result in derepression of the AVP-encoding gene and other targets of NRSF. Indeed, in NRSF-expressing cells, introduction of just the N-terminal repression domain (without an NRSE binding domain) appears to result in the loss of Sin3A, HDAC2, and hypoacetylated nucleosomes from the NRSE of the gene that encodes the M4 muscarinic receptor (21). This result is consistent with a model in which REST4 can sequester corepressor complexes away from full-length NRSF and thus indirectly derepress target genes (Figure 4⇓). Given that REST4 levels are elevated after kainate treatment (14), this mechanism may blunt NRSF-mediated downregulation of gene expression following status epilepticus.
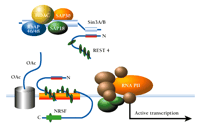
Proposed mechanism for activation of gene expression by derepression. Although NRSF is schematized to bind to the NRSE, the REST4 protein competes with NRSF for association with Sin3 corepressor complex. Thus sequestered away from interacting with NRSF, the corepressor–HDAC complex cannot remove acetyl groups (AcO) from nucleosomal core histones, and the RNA polymerase machinery (RNA PII) is thus able to form a transcriptionally active complex with the promoter.

Avtar Roopra, Ph.D., is currently a postdoctoral investigaor of Neurology at the University of Wisconsin. Yunfei Huang, Ph.D., is a postdoctoral investigaor of Neuroscience at Johns Hopkins University School of Medicine. Raymond J. Dingledine, Ph.D., is Professor and Chairman of the Department of Pharmacology at Emory University School of Medicine. Address correspondence to RJD. E-mail rdingledine{at}pharm.emory.edu fax 404-727-0365.
Although the majority of work on the NRSF–NRSE system has concentrated on the repression of gene expression, it has become increasingly clear over the years that the NRSE possesses enhancer activity as well as silencer activity. In a transgenic analysis of the gene that encodes the β2-nicotinic acetylcholine receptor (β2-nAChR), a point mutation introduced into the gene's NRSE resulted in loss of reporter gene expression in dorsal root ganglia and sympathetic ganglia (16). A transgenic study of the neuronally expressed gene that encodes the L1 cell adhesion molecule (17) similarly showed that disruption of the NRSE caused loss of expression in certain populations of neuronal cells, particularly in the hippocampus. Recently, NRSEs have also been implicated to enhance expression of the genes encoding dynamin (44), atrial naturitic peptide (24), and corticotropin-releasing hormone (45).
Although the mechanisms that determine whether an NRSE will act to silence or enhance gene expression have remained elusive, some inroads have been made. Foremost, the position of the NRSE within the gene appears to influence strongly how this element affects gene expression. In synthetic SV40-based reporter genes bearing an NRSE at different positions, a distal upstream NRSE repressed transcription, whereas one close to or downstream of the transcriptional start site activated transcription (16). That it was NRSF itself, rather than an unidentified NRSE binding protein, that mediated activation was established through NRSF-antisense knockdown experiments. The NRSEs that regulate β2-nAChR and L1 expression are downstream of their relative promoters, and are known to mediate transcriptional activation as well as repression (13, 16). Because repressor/activator function is thus determined as a function of genomic organization of target genes, NRSF may be able to repress some and activate other NRSE-containing genes simultaneously. The coupling of NRSF function to genomic organization thus economizes the cell's gene regulatory machinery.
How does NRSF activate transcription when bound to an NRSE downstream of the promoter? Either 1) the NRSF could recruit components of the basal transcription apparatus into an assembly that could extend to the promoter, or 2) the NRSF could indirectly affect promoter utilization by recruiting an HDAC that would deacetylate basal transcription factors rather than histones (46, 47). In both cases, the proximity of the NRSE to the promoter could be essential to activation. One can imagine a mechanism whereby a distal NRSE binds NRSF and depends on corepressor proteins to recruit or deacetylate promoter complex proteins provided that the promoter is topologically accessible. However, an NRSF–NRSE association located near the promoter may be able to recruit proteins to, or promote deacetylation of proteins at, the promoter in a manner either dominant to the influences of those recruited corepressors necessary for more distal promoters or mutually exclusive with such corepressor recruitment. That NRSF may work by helping to recruit basal transcription proteins is supported by the observation that a promoter-proximal NRSE can act synergistically with a distal enhancer to activate reporter gene expression in transient transfection assays (A. Roopra, unpublished observations).
Furthermore, direct effects of the NRSF system on the catalysis of transcription are worth considering. For example, the binding of NRSF or another protein to an NRSE located downstream of the promoter could facilitate RNA elongation, a key regulatory step in transcription. By comparison, the HIV TAT activator enhances transcription by overcoming an elongation pause (48, 49), and so it would be very interesting to assess whether NRSF exerts downstream activation by a similar mechanism. TAT requires a downstream element, TAR, to effect activation; however, unlike conventional transcriptional activators, TAT does not bind DNA, but rather binds the nascent RNA that contains the transcribed TAR sequence. Once transcribed, the TAR sequence forms a hairpin just behind the paused RNA polymerase complex. TAT, in turn, binds the hairpin and enhances elongation by recruiting the pTEFb complex and promoting subsequent phosphorylation of the RNA polymerase C-terminal domain. It will be interesting to see whether NRSF can also facilitate elongation by binding RNA that contains NRSE sequences.
CONCLUSIONS
The context in which NRSF functions has a major bearing on the role that this transcription factor plays. In addition to the issues described above that influence whether NRSF will activate or repress transcription, the complement of corepressors and other transcription factors present in the cell also has a decisive impact. The decision itself is dependent on cell type: activation is typically seen only in subpopulations of neurons, whereas NRSF–NRSE may be an obligate repressor in non-neuronal cells.
The two corepressors of NRSF, Sin3A and CoREST, have different spatio-temporal expression profiles through development (20). CoREST is found primarily in the head mesenchyme at around embryonic day 8.5 and becomes more widespread later in development, whereas Sin3A has a more ubiquitous expression pattern. This difference in availability of corepressors will likely determine NRSF's effect on NRSE-containing genes within a cell. It is likely that specific promoter architecture will also influence the activity of corepressor complexes. The ability of promoter architecture to influence repressor function has been documented for the Rb protein, which represses transcription by recruiting HDAC to E2F sites (50). However, a promoter can, if able to recruit Sp1, be rendered resistant to the HDAC arm of Rb repression (51). Sp1, as well as activating transcription by direct interaction with TAF130 (52), can bind acetylated histone H3 (53), an event postulated to prevent chromatin condensation mediated by deacetylation. One could thus hypothesize that NRSF may be less able to repress genes via the Sin3A/HDAC complex if Sp1 is present, but may still be able to utilize the CoREST complex if its repression mechanism is resistant to Sp1 chromatin effects.
Many transcription factors share a common corepressor, as demonstrated succinctly by Jepsen and colleagues (54), who deleted the NcoR corepressor by homologous recombination in mice. NcoR deletion resulted in the loss of function of multiple transcriptional repressors, including NRSF and Mad. The reliance on a limited number of corepressor complexes and a larger number of transcription factors allows for the intricate and sensitive crosstalk between seemingly disparate transcriptional repressors. For example, the transcriptional repressor PML interacts with a number of corepressors, including Sin3A and NcoR. During leukemogenesis, the formation of a PML–RAR chimera, by fusion of the two respective genes, generates a molecule with multiple SIN3 interaction domains. The alteration of PML–SIN3 interactions in cells harboring PML–RAR fusions, in turn, perturbs Mad–SIN3 interactions to the point that Mad repression is severely abrogated (55). Both Mad and NRSF interact with the N-terminal region of SIN3 (21, 22, 56), suggesting that interactions that disrupt SIN3 function affect Mad and NRSF alike. Thus, Mad and perhaps many other repressors, including NRSF, must be studied in the context of the larger transcription factor complement in the cell. A purely reductionist approach is insufficient to achieve a satisfying understanding of the roles of NRSF, an increasingly common theme in biology.
- © American Society for Pharmacology and Experimental Theraputics 2001

