PROMISCUOUS LIGANDS AND ATTRACTIVE CAVITIES
How do the inhaled anesthetics work?
Abstract
The inhaled anesthetics were officially introduced to American medicine more than 160 years ago and rank among the most important medical advances in our time. These drugs are used to render patients insensible over twenty million times each year and are the most dangerous of all drugs that physicians currently use. An entire medical specialty, anesthesiology, has arisen out of the need for the special training to administer them safely. Nevertheless, side effects, toxicity, and long-term cognitive problems continue to plague their use, especially in the very sick or aged. Hence, it is essential that we develop an understanding of their molecular pharmacology so that safer alternatives can be developed.

The scene depicted in Thomas Eakins' Agnew Clinic (see cover also) typifies surgery of the 1880s. An anesthetist delivers
diethyl ether from a can in his right hand and a paper cone over the patient's face. The enduring requirement for a burly
assistant to keep the patient from moving emphasizes the uncertain nature of anesthesia even at this advanced point in time.
Over 100 years after the scene was painted, a molecular understanding of anesthesia is far from complete. This Review discusses
some of the provocative concepts that inform current debates about anesthesia. [Agnew Clinic appears courtesy of the University
of Pennsylvania Art Collection, Philadelphia, Pennsylvania.]
WHAT IS ANESTHESIA?
In order to define a mechanism of drug action, it is useful to have measurable clinical endpoints that, ideally, are understood in terms of physiology and underlying pathways. So, what terms may be used to define “anesthesia”? In contrast to endpoints like blood pressure, heart rate, and temperature, behavioral states generally have vague, subjective definitions. Anesthesia is defined clinically as a state where no movement occurs in response to what should be painful (1). Although such a definition is convenient and necessary for pharmacological studies, it clearly does not describe the underlying neurophysiology. A more refined definition would consider anesthesia to be a composite of specific features like amnesia, hypnosis, analgesia, and immobility (2). But, similarly, the neurobiology of even these specific components is inadequately understood, and so the constellation of poorly understood features does little to offer insight. Nevertheless, these operational definitions can be taken together to characterize a reversible neurophysiological state in which a normally painful stimulus produces neither an outward response nor a memory.
Can a stimulus be painful if one neither reacts to nor remembers it? Because pain is strictly defined as the conscious interpretation of noxious stimuli, the answer must be no. But any anesthesiologist knows that physiological manifestations of pain occur in otherwise well-anesthetized patients. So, is analgesia an essential component of anesthesia? Is "anesthesia" (i.e., the absence of sensation) the proper term to describe a condition that is more accurately characterized by the inability to interpret, respond, or remember pain? Is anesthesia the result of CNS depression, even though most inhaled anesthetics have a well-known excitatory component?
These questions demonstrate that a poorly defined endpoint, and an absence of a reliable in vitro correlate, considerably hinder the determination of drug mechanism, especially when the drug primarily affects consciousness, arguably the least understood aspect of biology. Will the identification of the molecular target(s) of anesthetics help elucidate the physiological mechanism of elicited unconsciousness_and thereby help to define consciousness itself? The possibility of establishing the physiological basis of consciousness is but one motivation that drives research into anesthetic mechanisms of action.
So, what do we really know about how the inhaled anesthetics produce insensibility? These drugs were introduced during an age of invention, the latter nineteenth and early twentieth centuries. Because successful inventions were often rewarded in tangible ways, many different volatile compounds were tried on patients and animals (often, first on the inventor), and many produced the desired state, albeit with widely varying potency. For example, a mere whiff of chloroform could be sufficient to produce unconsciousness, whereas the anesthetic action of diethyl ether or cyclopropane required much more vapor. Nitrous oxide was particularly impotent, being required at pressures over a full atmosphere.
In the early twentieth century, Overton and Meyer independently noted that the more potent drugs were also more soluble in olive oil, and researchers soon verified an impressive correlation between anesthetic potency and oil solubility (Figure 1⇓) (3). This early work predated our understanding of the composition and structure of the cell membrane and proteins, so conclusions were limited to the probability that anesthetics produce their effect by acting on undefined fatty components of the cell.
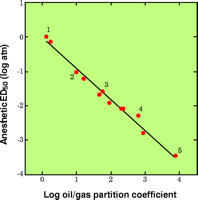
The lipophilicity of inhaled anesthetics. At the turn of the century, Meyer and Overton independently noted the strong correlation between the oil solubility of inhaled gases and their anesthetic potency. Of twelve inhaled agents analyzed, five are identified: 1, nitrous oxide; 2, cyclopropane; 3, diethyl ether; 4, trichloroethylene; 5, thiomethoxyflurane.
CHEWING THE FAT: LIPIDS AS ANESTHETIC TARGETS
With the discovery that biological membranes are constructed largely of lipid and are, therefore, olive oil–like, a logical extension of Overton and Meyer's work was the hypothesis that the inhaled anesthetics act by targeting the cell membrane. Moreover, because important cell signaling pathways depend on proteins embedded in the cell membrane, the dissolution of sufficient lipophilic agents, such as the inhaled anesthetics, was predicted to alter some important physical property of the lipid bilayer, which in turn would change the function of embedded proteins. This “unitary hypothesis” launched a host of studies that indeed showed the inhaled anesthetics to change lipid bilayer properties (4). These changes, however, tended to be small, and were detectable only at anesthetic concentrations manyfold higher than those necessary to produce anesthesia (5).
But how much change in the biophysical properties of lipid bilayers is necessary to appreciably alter cell signaling? One approach to address this question comes from phase transition studies of model membranes (essentially, analysis of their melting points) under varied conditions. Specifically, clinical concentrations of inhaled anesthetics decrease lipid phase transitions by 1–2oC (6), a perturbation that would be analogous to that expected from an elevation in body temperature of similar magnitude. Because low-grade fevers do not produce insensibility, this finding casts doubt as to whether unitary lipid mechanisms could accurately explain anesthesia. Similarly, some compounds that fail to induce anesthesia can nevertheless mimic anesthetics in perturbing lipid phase transitions (7). The final blow, in the eyes of some researchers, to the hypothesis that the inhaled anesthetics act predominantly on lipid bilayers, was delivered by the finding that stereoisomers of a commonly used anesthetic, isoflurane, have an identical effect on lipid phase transition despite manifesting a significant difference (~20%) in their in vivo potencies (8,9).
Although many have viewed such evidence as the coffin nails in lipid-centered theories, several caveats must be considered before eliminating membrane lipid as an important direct target for inhaled anesthetics. First, most studies have used nonbiological lipid systems as models. Dipalmitoylphosphatidylcholine (DPPC) for example, being a major component of egg lecithin and having no double bonds to oxidize, is easy to obtain and work with. DPPC molecules, furthermore, pack together nicely into highly organized gel states that undergo dramatic, easily measured phase transitions into the biologically relevant liquid crystalline state. Although the experimental convenience of DPPC molecules largely account for their widespread use in the laboratory, they fail to faithfully mimic cell membranes, especially those of neurons, in several respects. Biological membranes, for instance, are highly heterogeneous mixtures of lipids (saturated and unsaturated, charged and neutral) and cholesterol. In addition, proteins account for approximately 50% of the mass of the cell membrane. These biological realities (Figure 2⇓) result in cell membranes that, unlike model DPPC systems, do not exhibit clear phase transitions, and thus the modulation of model lipid phase transitions may be irrelevant when it comes either to confirming or refuting lipid membranes as anesthetic targets.
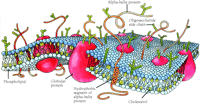
Schematization of the plasma membrane, showing phospholipids, cholesterol, sugars, and various proteins. Note the complexity apparent even in this extremely simplified rendering. [Reproduced with permission from Scientific American 253, 100-108 (1985).]
Second, membranologists are only beginning to understand the complexity of membranes and the subtle but crucial coupling between membrane proteins and the lipid bilayer in cell signaling mechanisms. We now know that the inner and outer leaflets of the bilayer are often distinct in lipid composition, and that certain lipids and proteins are organized laterally into islands, or rafts, of unique (and chiral) composition (10), all of which may strongly influence protein activities in vivo. For example, the activity of rhodopsin, a G protein–coupled receptor that transduces photons in the visual system, is functional only when a significant fraction of the membrane phospholipid contains a specific fatty acid (11), whereas the nicotinic acetylcholine receptors in nerve and muscle cells need cholesterol and anionic phospholipid (12). Other membrane proteins are functionally dependent on lipids with specific charged head groups. Thus, homogeneous lipid systems cannot begin to approximate the complexity of biological membranes. Without a clear understanding of the mechanisms of lipid–protein coupling, the precise roles of lipid components that contribute to anesthesia remain unclear.
PUTTING ON THE SQUEEZE: A NEW LIPID HYPOTHESIS
A recently proposed lipid-centered mechanism of anesthesia relies on a fundamental property of amphiphilic molecules, such as phospholipids, as they associate, in aqueous medium, into bilayers. Specifically, a distribution of non-uniform lateral pressure values arises within the bilayer (Figure 3⇓), which could influence the conformation of integral membrane proteins so that they “sense” the “pressure status” across the lipid bilayer (13). In addition to providing a possible coupling mechanism between lipid and protein, the theoretical lateral pressure profile would be dependent upon the specific compounds that distribute across the bilayer heterogeneously. Most evidence confirms that anesthetics do indeed distribute unevenly across the bilayer, concentrating in more amphiphilic regions as compared to the purely hydrophobic membrane interior (14,15). The theory certainly merits further investigation. A prediction, for example, is that long stiff hydrophobic molecules should be less potent anesthetics than more flexible congeners.
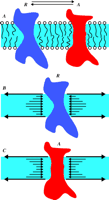
Lateral pressures within the membrane may control membrane protein conformational equilibria. Panel A shows the equilibrium between two conformational states (R for resting, and A for activated) of a hypothetical membrane protein. Panels B and C use arrows to represent the direction and magnitude of lateral pressure in a phospholipid membrane; the surface tension at the phospholipid/water interface is balanced by pressure of the opposite direction in the hydrophobic “core” of the membrane. The pressure in this core region is not constant across the bilayer; panel B shows that excess pressure in the middle will favor the R conformation of the protein, whereas the pressure profile represented in panel C will favor the A conformer. Changes in this lateral pressure profile are therefore expected to influence membrane protein conformational and insertion equilibria.
The lateral pressure idea can also be tested via theoretical calculations. With the availability of reliable, high-speed computer models of both saturated and unsaturated phospholipids, researchers should be able to use molecular dynamic simulations to compute lateral pressure profiles and thereby predict anesthetic effects. So far, computer simulations are consistent with an asymmetric distribution of inhaled anesthetics across the bilayer (Figure 4⇓) and predict a surprisingly large effect on orientation of the phosphocholine dipole (16). Such surface electrical properties could have profound effects on certain membrane proteins—on voltage gated ion channels, in particular—and could provide additional anesthetic mechanisms based on coupling between membrane proteins and the lipid bilayer.
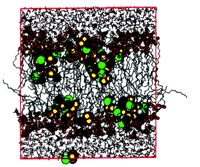
A proposed anesthetic mechanism that includes contributions from both the lipid bilayer and membrane proteins. Halothane (consisting of the larger, colored spheres), a typical inhaled anesthetic haloalkane, localizes preferentially to the amphiphilic regions (shown as small, colored spheres) of the bilayer (in which the acyl side chains are shown as ball-and-stick strands) in this molecular dynamics simulation; the aqueous milieu is represented at the upper and lower peripheries of the red box. [Reproduced with permission; Koubi, L. et.al., Biophysical Journal 78, 800–811 (2000).] This non-uniform distribution makes it likely that the lateral pressure profile will be altered.
Thus, lipocentric hypotheses, although currently not favored, cannot yet be discarded. But to formulate reasonable hypotheses, we need first to better understand how lipids and proteins interact. We must also develop experimental and theoretical tools for measuring these interactions so that lipocentric hypotheses can be adequately tested.
DO PROTEINS HOLD THE KEY?
Proteins currently hold the monopoly as favored molecular targets in theories of anesthesia. The concept that direct interactions between anesthetic and protein might underlie anesthesia is neither new—Claude Bernard proposed it in 1875—nor, to the extent that proteins contain hydrophobic domains to which small apolar molecules can gain easy access (17,18), inconsistent with the Overton–Meyer correlation. But protein-based hypotheses gained significant momentum when it was established that anesthetic pharmacodynamics could be mimicked in lipid-free systems. For example, most anesthetics inhibit the activity of luciferase—a soluble enzyme—in a manner that correlates with their in vivo anesthetizing potency (Figure 5⇓) (19). This finding set the stage for the next major phase of investigations into anesthetic mechanisms, namely, the proteocentric phase.
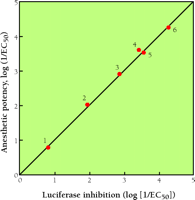
Correlation between the inhibitory activity of several volatile anesthetics with respect to firefly luciferase and in vivo anesthetic potency. Note the remarkable correlation over four orders of magnitude, validating the use of surrogate proteins to characterize anesthetic targets. 1, acetone; 2, diethyl ether; 3, chloroform; 4, halothane; 5, methoxyflurane; 6, n-pentane. [Data from Franks, N.P. and Lieb, W.R. Nature 310, 599 (1984).]
For decades, the modus operandi for pharmacological research has been to posit the existence of a protein receptor for a ligand of interest. The usual pharmacological search starts by analyzing receptor–ligand binding characteristics. Tissues, cells, or membranes are probed with radiolabeled ligands so as to identify high-affinity binding sites, which are then progressively characterized to determine what the “receptor” actually is. According to this standard protocol, analysis of how the ligand modulates the function of identified receptors allows one to determine their physiological roles. But this approach was technically prohibitive in the search for receptors of the inhaled anesthetics. These ligands are small, volatile, and not generally amenable to assay. Clinical EC50 values (the plasma concentration resulting in 50% of a population being anesthetized) are in the low millimolar range (20), which strongly suggests the absence of high-affinity binding and relegates the duration of ligand–receptor interactions to a millisecond or less. Conventional binding assays simply won't work for such low-affinity binding.
Given the difficulty of identifying direct interaction between anesthetics and protein receptors, laboratories began to investigate the molecular pharmacology of anesthetics by measuring their effects on the activity of various in vitro preparations and by using “plausibility” and “sensitivity” as the two principle criteria for judging relevance to anesthetic action. “Plausibility” means that modulation of a given target's activity fits our preconceptions of an anesthetic mechanism. In other words, because anesthesia is often thought to represent depression of central nervous system activity, either inhibition of an excitatory neurotransmission system, or activation of an inhibitory one, fits the plausibility criterion. If experiments show an anesthetic to affect both classes of neurotransmission—as they generally do—then “sensitivity” can distinguish which is the more important. For purposes of simplification, the sensitivity criterion is satisfied by that in vitro effect (inhibitory or excitatory) with an anesthetic EC50 closest to clinical EC50 values.
Admittedly, given that we don't really understand the neurophysiology of consciousness, plausibility is in the eye of the investigator. The inhaled anesthetics have been applied to numerous in vitro systems, and quite remarkably, they alter the function of many. Enzymes, receptors, carriers, transporters, both ligand- and voltage-gated channels, and even structural proteins have been modulated in some way by inhaled anesthetics (21). Clearly, plausibility alone does not provide much discrimination, but in concert with the sensitivity criterion, some degree of target specificity is discernible. Although most effects of anesthetics are observed within a fairly narrow range of concentration (~0.2 to 5 mM), the ligand-gated ion channels, especially the inhibitory ones, appear to be more sensitive than most of the other proteins implicated by the plausibility criterion alone (22).
SURFING THE CHANNELS
The implication of inhibitory ligand-gated channels in anesthesia is compelling. These receptors are coupled to chloride channels, so that upon activation by an agonist (e.g, γ-aminobutyric acid), chloride flows into the cell, hyperpolarizing the membrane and reducing the likelihood of signal propagation. Drugs like diazepam (Valium) are thought to reduce anxiety by potentiating the activity of inhibitory channels, and other hypnotics, like the barbiturates, may act similarly. It is therefore plausible that inhaled anesthetics would have similar effects. Indeed, clinical concentrations of inhaled anesthetics significantly potentiate chloride currents that are activated by either γ-aminobutyric acid (GABA) or glycine in various in vitro models (23,24). But ambiguities lurk. For example, the inhibitory ligand-gated ion channel model would predict excitation of the central nervous system at high anesthetic concentrations because GABAA activity tends to be inhibited at greater than 1 mM anesthetic. In practice, however, high concentrations of anesthetics only deepen anesthesia, with no apparent discontinuity. In addition, although GABA is an excitatory transmitter in the rodent newborn, where chloride gradients are reversed, the inhaled anesthetics still cause insensibility, albeit at slightly higher concentrations (25).
There are other nagging doubts regarding the identification of inhibitory ligand-gated channels as the targets of inhaled anesthetics. If the anxiolytic action of the benzodiazepines (Valium-like drugs) is due to the potentiation of GABAA receptors, then inhaled anesthetics clearly must act differently, because they are distinctly non-anxiolytic, at least at concentrations where patients can still report their state of mind. During the induction of anesthesia, in fact, most of the inhaled anesthetics cause an “excitement” stage, in which patients unconsciously flail about. Some inhaled agents even cause seizure-like activity, whereas diazepam and its analogs prevent seizures. Further, chloride channel blockers, and GABAA receptor antagonists in particular, have only small effects on the potency of inhaled anesthetics (26,27), and interestingly, genetic knockout of the GABAA receptor, although often lethal, only minimally alters anesthetic sensitivity in survivors (28,29). Of course, gene knockout experiments, often complicated by the problem of developmental adaptation to the absence of important signaling systems, may result in ambiguous data. Such studies nevertheless permit the conclusion that the GABAA receptor does not constitute a unique receptor for the inhaled anesthetics. Thus, although dramatic potentiation of the inhibitory ligand-gated channels exists under some conditions in vitro (and probably in vivo), it remains unclear how this potentiation contributes to anesthetic action.
PROMISCUOUS LIGANDS: STRENGTH IN NUMBERS
So, do interactions with more than one molecular target underlie inhaled anesthetic action? Probably. This multiplicity is suggested not only by effects on a wide variety of proteins, but also by the wide variety of different molecules that can produce anesthesia. From the monoatomic gas xenon to the halogenated branched ethers (Figure 6⇓), all result in anesthesia when inhaled at the right concentration. How could all of these low-affinity compounds interact in a specific way with a single receptor? Some recent evidence suggests a unique binding site for each of several inhaled anesthetics on the same molecular target (30,31), but the likelihood of only one protein target having specific sites for all of the small molecules tested seems remote. Other recent evidence suggests a unique molecular target for each of several classes of anesthetic (32,33), but again, such selectivity seems extraordinary. At the least, the notion of multiple selective targets opens the door for multiple pathways to the final effect, anesthesia. It is only another small conceptual step to view actions of a single anesthetic at each target contributing to its final effect.
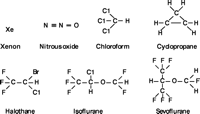
Chemical structures of seven inhaled anesthetics. Two are no longer used clinically: chloroform, due to its toxicity; and cyclopropane, because it is explosive. Note the small differences in structure and size, small permanent dipole, and the lack of aromatics or good H-bond acceptors and donors. The more potent anesthetics (e.g., halothane) tend to have more polar functions.
Do comparative population studies provide any discrimination between multiple and single target models? The dose-response curve for inhaled anesthetics within a human population is very steep (Figure 7⇓), indicating low variability of responsiveness to anesthetics in the population (34). Drugs known to act via single molecular targets, on the other hand, result in broader dose-response curves when administered to a population, reflecting receptor polymorphisms and other sources of variability. Responsiveness to the inhaled anesthetics, moreover, is surprisingly conserved across the entire animal kingdom, where large differences in general receptor composition otherwise exist. These drugs even have effects in plants! Only slightly higher inhaled anesthetic concentrations are required to prevent movement in mimosa plants than in humans (35). Further, significant resistance to the inhaled anesthetics is uncommon, tends to occur in simple creatures (e.g. nematode, yeast), and has not been convincingly produced by mutagenesis or selective breeding. Interestingly, mutagenesis has produced sensitive organisms, the genetic analysis of which has suggested multiple contributing targets (36). Finally, tolerance, a relatively common feature of drugs that act via unique receptors, has not been demonstrated for the inhaled anesthetics. Thus, features within and between populations of organisms would appear to point away from highly selective targets for the inhaled anesthetics.
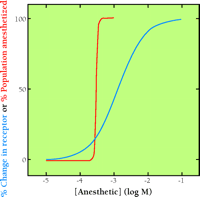
Typical dose-response curves for a) in vivo anesthesia (red), and b) a typical in vitro response to activation of a single receptor (blue). Note the much steeper curve for anesthesia (Hill coefficient ~20) as compared to a typical receptor curve (Hill coefficient of ~1). Even highly cooperative enzymes or receptors rarely show Hill coefficients greater than about 3 for ligand binding.
At least two potential explanations exist for the observations listed above. First, a unique “anesthesia receptor” could be essential to survival, such that its anesthetic binding domains are well conserved. This essentiality could explain knockout results, too, in that the animals with a true absence of this important receptor simply don't survive. The second explanation could be that multiple targets underlie anesthetic action, each contributing only a fraction of the anesthetic effect. Thus, variability in any single target would be difficult to detect, and the development of resistance would be minimal without multiple mutations. Further, although the mechanisms of tolerance are not completely clear, it is reasonable to speculate that any given receptor response is less likely to induce tolerance if only minimally affected by anesthetics in the first place.
But what about stereoselectivity (8)? Surely, if the targeting of each of a very large number of protein targets makes a small contribution to net anesthesia, individual steric selectivity for ligands should be statistically obliterated. Yet, for at least one anesthetic, isoflurane, stereoselectivity has been reasonably well established in vivo (9). The difference in potencies between the stereoisomers, however, is small (about 20%), and without knowing either the number or the range of selectivity of individual targets, an assessment of the role of stereoisomerism in establishing potency is difficult. It is not enough to identify a target with selectivity that matches in vivo selectivity; many such proteins (some clearly implausible) have satisfied this criterion (37,38). Such a small degree of stereoselectivity may even arise from the chirality of natural amino acids and backbone structure in proteins. Thus, although generally viewed as implicating protein targets, stereoselectivity cannot eliminate a multiple target model.
Models of specific, direct protein action on the one hand and multiple targets on the other are not mutually exclusive; ATP, oxygen, and calcium are clear examples of ligands that interact with multiple proteins. Inhaled anesthetics, too, bind specifically to many proteins: firefly luciferase (39), serum albumin (40), myoglobin (41), adenylate kinase (42), haloalkane dehalogenase (43), and T4 lysozyme (44). Although only one meets the plausibility criterion (i.e., adenylate kinase), these data clearly show that anesthetics can bind specifically to multiple targets. Even more compelling evidence comes from recent anesthetic binding experiments in brain slices (45). Photolabeling with a radiolabeled halothane derivative revealed widespread binding of halothane throughout the brain (Figure 8⇓), the distribution of which does not match that of any known receptor or channel. When extracted and separated by electrophoresis, dozens of brain proteins were specifically labeled (in a saturable and stoichiometric manner), with estimated affinities close to the clinical EC50 (46). Hence, the inhaled anesthetics can indeed be promiscuous ligands. But this should not be surprising. The clinical EC50 for these drugs, which is in the millimolar range, is 103–106 times higher than drugs that are known to act on single receptors. Indeed, their small surface area, hydrophobicity, and generally unreactive nature predict the weak and non-stringent binding of the inhaled anesthetics.
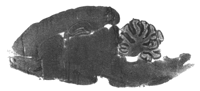
Autoradiogram of rat brain section photoaffinity labeled with radioactive halothane. Degree of halothane binding is indicated by level of darkness; no other staining has been applied to the section. The binding shows little regional preference and is reduced to near-background levels in the presence of a tenfold excess of unlabeled halothane. The dramatic inhibition of labeling by non-radioactive halothane indicates that most halothane binding is saturable and specific, showing that many proteins could be involved in anesthetic action [from (45)].
ATTRACTIVE CAVITIES: FILLING IN THE HOLES
How can the weak binding that is manifested by the inhaled anesthetics elicit such profound biological effects? As a start, we need to understand how these drugs interact with proteins. It is well-known that proteins fold into complex structures with packing defects termed “cavities.” Cavities are thought to be necessary for protein function, since they introduce instabilities that facilitate conformational changes (47,48). Although most protein cavities are too small to accommodate anesthetic molecules, many proteins—and particularly those embedded in membrane—possess cavities of capacities appropriate for the binding of anesthetics. Within the hydrophobic protein interior, such cavities are attractive sites for the binding of hydrophobic anesthetics (Figure 9⇓) in a manner consistent with the Overton–Meyer correlation. The fact that potent anesthetics appear to possess a small degree of polarity is also consistent with an affinity for protein cavities, as most are weakly polar. Further, backbone atoms form a portion of the lining of many cavities, which is relevant because protein backbones manifest chirality and generally conserved secondary structures. Thus, a small degree of stereoselectivity could be consistent with multiple protein targets.
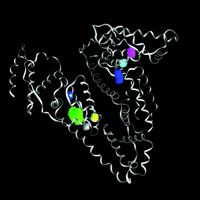
Structure of human serum albumin [Protein Data Bank #1AO6 (40)], showing the carbon backbone as ribbons, and several internal cavities. These cavities are depicted as solids, representing the cavity surface, with color used for distinction. Only the green cavity is sufficiently large to bind a typical inhaled anesthetic without distortion of the native protein structure.
How does occupancy of pre-existing protein cavities by small organic anesthetic molecules invoke such a profound effect on behavior? One possibility is that stabilization of the protein, through occupancy of a cavity, reduces the motion that underlies normal protein activity. A number of different methods have indeed verified that occupancy of internal cavities by small hydrophobic molecules reduces protein motion (e.g., as measured by stability or by crystallographic B-factors) (47,49). This “calm molecules, calm minds” hypothesis has an attractive simplicity. It is also clearly a multiple-target hypothesis, nicely reconciling binding and functional results.
On the other hand, the anesthetic-induced activation of inhibitory ligand-gated channels (see above) may at first appear at odds with the “calming” of protein function through an interaction between anesthetic molecules and protein cavities. Can a receptor be both “calm” and activated? To answer this apparent contradiction, it is necessary to consider the concept of conformational ensembles. The standard concept of drug action is that, subsequent to binding, a protein receptor assumes an altered conformation that possesses altered functionality. This view, however, is probably inaccurate. It is more likely that a given protein is always populating, to a widely varying degree, a large ensemble of different conformations (Figure 10⇓). Only certain pre-existing conformations, with optimal features for drug binding, will be stabilized by the drug, so that, by mass action, the population of protein molecules in the drug-binding conformation—that is, the conformation that possesses altered functionality—will grow. Thus, anesthetics could either enhance or inhibit protein activity, depending on which conformer in the ensemble contains the most attractive cavities. Is it possible to use the attractive cavity concept to estimate the number of anesthetic targets? At this point, the answer is a very limited yes. For example, using only cavity volume as a criterion, we find (Figure 11⇓) that about 15% of the high-resolution protein structures in the PDB protein data bank have internal cavities large enough to accommodate typical inhaled anesthetics (~150Å3). (This estimate assumes binding only to the crystallized conformer.) Further, most of the PDB structures are of the more easily crystallized soluble proteins, and not the membrane proteins that are the putative anesthetic targets and that have larger cavities (50). Nevertheless, this simple analysis suggests anesthetics bind only a subset of proteins, which should facilitate the assessment of the pharmacologically relevant proteins. But whether all these binding targets contribute functionally to the state of anesthesia remains an important and unanswered question.
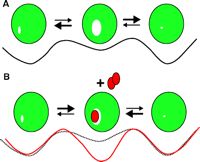
Cartoon of a protein (green circles) conformational ensemble with hypothetical free-energy curves depicted underneath. A. The middle conformation has an enlarged internal cavity, at the expense of a higher free energy, and is therefore poorly populated at equilibrium as shown by the size of the arrows.B. The addition of an anesthetic (red ovals) that binds to the cavity preferentially stabilizes and favors the central conformer. Whatever activity is associated with this conformer (e.g., activation of chloride channels) would therefore be enhanced by the anesthetic.
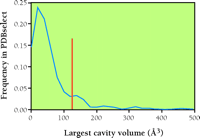
A group of ~500 soluble proteins in the PDB (PDBselect) were analyzed for the presence and volume of internal cavities, and this figure shows the results for the largest cavity found in each. Note that most proteins only have small cavities (~40-50 cubic angstroms), but around 10% have internal cavities which could fit typical inhaled anesthetics (~150 cubic angstroms), as indicated by the red line. Note that this simple analysis does not include membrane proteins that generally have larger cavities, nor does it include open hydrophobic pockets on the proteins, which may also bind anesthetics, albeit more weakly.
CONCLUSIONS
Discovery of an anesthetic receptor would indeed be important and dramatic, and is for many reasons a doggedly pursued goal. However, taken together, the above considerations seem to point in another direction, namely, that the integrated, final common pathway that we term “anesthesia”, is due to small effects at many biological sites. Such a mechanism of drug action is poorly represented in the pharmacology literature, which remains entrenched in the single-target model for drugs. A small-effects-at-many-sites model is therefore considered inelegant, because it will be difficult to validate. Perhaps also because it calls for a skill-set unfamiliar to many pharmacologists—integrative biology—to assemble the multiple molecular effects into an organismal outcome. Nevertheless, this multiple target hypothesis is most consistent with clinical observation, the available data, and the remarkable resistance that this 160 year-old dilemma has offered to solution.
Acknowledgments
Funding for the author's work is provided by NIGMS 51595 and 55876.
- © American Society for Pharmacology and Experimental Theraputics 2001
References

Roderic G. Eckenhoff, MD, is Austin Lamont Professor of Anesthesia and Director of Research at the Department of Anesthesia at the University of Pennsylvania. He is a practicing anesthesiologist, and heads a multidisciplinary team that investigates mechanisms of anesthesia. E-mail roderic.eckenhoff{at}uphs.upenn.edu; fax 215-349-5078.

