Fractalkine/CX3CL1: A Potential New Target for Inflammatory Diseases
Chemokines are small, chemoattractant proteins that recruit inflammatory cells at the site of inflammation. Because of this, chemokines and their receptors are considered to be therapeutic targets in chronic inflammatory disorders (1). Humans have more than fifty distinct chemokines, small (8–10 kDa) heparin-binding proteins that were identified by their chemotactic activity in bone marrow–derived cells (2). Broadly based on the findings, two types of chemokines exist: 1) inflammatory chemokines, which recruit leukocytes in response to physiological stress, and 2) homeostatic chemokines, which account for basal leukocyte trafficking and the formation of the architecture of secondary lymphoid organs (3, 4). Inflammatory chemokine expression can be elicited by almost any stimulus that alters cellular homeostasis, such as infections and immune disorders (4).
Chemokines are categorized into four subfamilies according to the number and spacing of the first two cysteines in a conserved cysteine (C) structural motif (5). The four subclasses are: C, CC, CXC, and CX3C. The chemokine receptors are bound to the cell membrane through seven-transmembrane helical segments coupled to a G protein that transduces the intracellular signal. The two major subclasses that emerged from this classification include the CC chemokines, where cysteines are adjacent, and CXC chemokines, where the cysteines are separated by one amino acid. Receptors were designated as CXC, CC, C, and CX3C followed by “R” and a number (e.g., CXCR5), and chemokines were assigned the same acronyms followed by “L” for ligand type (5). The CXC chemokines, such as IL-8/CXCL8, act mainly on neutrophils and lymphocytes, whereas the CC chemokines, such as MCP-1/CCL2, act mainly on monocytes and lymphocytes without affecting neutrophils (6). Chemokines in endothelial cells and their respective receptors on inflammatory cells play a critical role in cellular recruitment and retention, and resolution of inflammation (6).
Most chemokines are synthesized as proteins in sequences of 20–25 amino acids. The mature form of some chemokines is secreted and processed to yield truncated forms with various levels of biological activity (7). Because the N-terminal sequence preceding the first cysteine is exposed and conformationally disordered, many chemokines are easily processed at the N terminus. This proteolytic cleavage amplifies the biological activity of CXC inflammatory chemokines (7). Only a few studies have reported chemokine processing at the C terminus or at internal sites. Generally, the removal of up to six C-terminal amino acids has little to no effect on their chemotactic activity (7).
In the late 1990s, researchers discovered a unique chemokine subclass, differing from the known chemokines (8). The CX3C chemokine subclass contains only one member, termed fractalkine or CX3CL1 (5, 9). Fractalkine/CX3CL1, composed of 373 total amino acids, is also unique among chemokines because it is synthesized as a transmembrane molecule consisting of an extracellular N-terminal domain (residues 1–76), a mucin-like stalk (residues 77–317), a transmembrane α helix (residues 318–336), and a short cytoplasmic tail (residues 337–373) (10) (Figure 1). The soluble form consists of the chemokine domain and the extracellular mucin-like stalk, while the membrane-bound form functions as an adhesion molecule and promotes shear-resistant adhesion of CX3CR1 leukocytes. Soluble fractalkine/CX3CL1, which has a chemoattractive function, is generated by disintegrin-like metal-loproteinases ADAM 10 and ADAM 17/TACE (11, 12). It is believed that ADAM 10 is responsible for constitutive shedding, whereas ADAM 17 activity enhances cleavage in response to cell activation. The soluble form has chemoattractive activity for monocytes, natural killer (NK) cells, and T cells (8). Membrane-bound fractalkine/CX3CL1, which supports integrin-independent leukocyte adhesion, can be induced on primary endothelial cells by inflammatory cytokines such as TNF-α, interferon-γ (IFN-γ), and interleukin-1 (IL-1) (13).
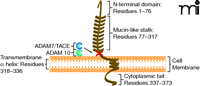
Membrane-bound fractalkine/CX3CL1. Schematic structure representation of fractalkine/CX3CL1, a membrane-bound chemokine consisting of a extracellular N-terminal domain (residues 1–76), a mucin-like stalk (residues 77–317), a transmembrane α helix (residues 318–336), and a short cytoplasmic tail (residues 337–373) (10, 57). Membrane-bound fractalkine/CX3CL1 is proteolytically cleaved by ADAM 10 and ADAM 17/TACE to generate soluble fractalkine/CX3CL1, which consists of the chemokine domain and the mucin-like stalk (11, 12, 58). The soluble form has chemoattractive activity for monocytes, natural killer cells, and T cells (9).
CX3CR1 is a highly selective chemokine receptor and surface marker for cytotoxic effector lymphocytes––such as NK cells, cytotoxic T lymphocytes (CTLs), and γδ cells––which express great amounts of perforin and granzyme B, regardless of their lineage and mode of target cell recognition (10). In the peripheral blood and synovial fluid (SF), fractalkine/CX3CL1 is expressed in monocytes during inflammatory conditions. Fractalkine/CX3CL1 is also expressed in macrophages, fibroblasts, endothelial, and dendritic cells in rheumatoid arthritis (RA) synovial tissue (14). Presence of fractalkine/CX3CL1and its receptor CX3CR1 in lymphocytes and their enhanced cellular expression during inflammatory conditions suggests that the CX3CL1-CX3CR1 duo participates in immune-related inflammatory diseases.
The classical pathway of leukocyte migration starts with selectin-mediated interactions between rolling leukocytes and the endothelium. Integrins on leukocytes are activated by locally produced chemokines that are presented on glycosaminoglycans (GAGs), triggering adhesion between leukocytes and endothelial cells. The leukocytes are then brought into the vascular wall and surrounding tissue (10). Before fractalkine/CX3CL1 was discovered, it was thought that all chemokines were secreted as soluble molecules that associate with cell surface proteoglycans and tissue matrix components (such as GAG) to maintain the local chemokine gradient (15). After association, the interaction between chemokines and their specific receptors on leukocytes activates members of the integrin family of adhesion molecules through a mechanism involving G protein–coupled receptors (3, 4).
When fractalkine/CX3CL1 is present, the mechanism for leukocyte adhesion is altered. The chemokine domain is presented at the top of the membrane-bound, mucin-like stalk, where it acts as an adhesion molecule. The fractalkine/CX3CL1 adhesion mechanism makes the association with proteoglycans and other adhesion molecules unnecessary. Immobilized fractalkine/CX3CL1 exhibits rapid and high affinity binding to CX3CR1-expressing cells in both static and physiological flow conditions (10). Interaction between fractalkine/CX3CL1 and CX3CR1 can markedly increase integrin affinity, which results in much firmer adhesion (16). Leukocytes then extravasate through the vascular wall and into the tissue to a chemokine gradient. Fractalkline/CX3CL1 may enhance extravasation of leukocytes by mediating cellular adhesion through the initial tethering and transmigration steps (10) (Figure 2).
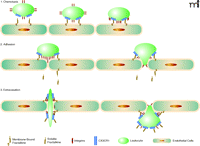
Fractalkine/CX3CL1: Sequential steps involved of chemotaxis. Schematic mechanism of fractalkine/CX3CL1 chemotaxis. When fractalkine/CX3CL1 is present, the chemokine domain is presented at the top of the membrane-bound mucin-like stalk, where it acts as an adhesion molecule. Immobilized frac-talkine/CX3CL1 exhibits rapid and high-affinity binding to CX3CR1 (10). Interaction between fractalkine/CX3CL1 and CX3CR1 can increase integrin affinity, which results in firmer adhesion (16). Soluble fractalkine/CX3CL1 binds to CX3CR1, aiding leukocyte extravasation through the vascular wall and into the tissue (10).
The association between CX3CR1 and integrins through the coexpression of fractalkine/CX3CL1 and integrin ligands, such as intercellular adhesion molecule (ICAM)-1 and vascular adhesion molecule (VCAM)-1, greatly enhances cell adhesion function compared with each system alone (10). Studies have suggested that fractalkine expression at the site of inflammation can attract and activate NK cells, resulting in the consequent lysis of neighboring endothelial cells (17). Fractalkine/CX3CL1 expressed on inflamed endothelium may play a role as a vascular gateway for cytotoxic effector cells (CX3CR1-expressing cells) by rapidly capturing them from the blood and promoting migration into tissue (10).
Atherosclerosis is a common condition that ranges from asymptomatic disease to stable angina pectoris to acute coronary syndrome (ACS), including unstable angina pectoris (UAP). Atherosclerotic lesions contain large numbers of immune cells (macrophages and T cells) (18). Chemokines aid in monocyte migration to the vessel wall where they act as precursors to foam cells (i.e., vessel wall–localized aggregates of macrophages and smooth muscle cells that have accumulated low-density lipoproteins) in atherosclerosis (19). Infection-induced production of inflammatory cytokines, such as TNF-α, IFN-γ, and IL-l, may stimulate fractalkine/CX3CL1 production in endothelial cells, contributing to atherosclerotic plaque development and rupture (10, 18, 19). Fractalkine/CX3CL1 expression is increased in athero-sclerotic lesions of apolipoprotein E-deficient mice, and crossing CX3CR1−/− with apoE−/− decreases atherosclerotic lesion formation with reduced macrophage accumulation (10, 20, 21).
Fractalkine/CX3CL1 also appears to mediate pathophysiological processes in cardiovascular disease (CVD). Ikejima et al. found increased expression of fractalkine/CX3CL1 and CX3CR1 in monocytes, T lymphocytes, and NK cells in ruptured coronary plaques in patients with UAP (22). The amounts of soluble fractalkine/CX3CL1 were substantially increased in patients with plaque rupture as compared to concentrations of the chemokine in patients within other groups (22). Possible mechanisms for the role of frac-talkine/CX3CL1 in coronary plaque rupture include an increase in CX3CR1-expressing monocytes or increased macrophage activation by CX3CR1-expressing T lymphocytes and NK cells at the site of inflammation (22). Luizzo et al. reported that monocytes in UAP patients show IFN-γ signal activation, suggesting that monocyte activation by IFN-γ is stimulated by NK cells and T lymphocytes (23). These findings suggest that increased CX3CR1-expressing monocytes, T lymphocytes, and NK cells may result in a burst of macrophage activation that could lead to plaque rupture (22). Furthermore, White and coworkers showed that fractalkine/CX3CL1 has proliferative and anti-apoptotic effects on primary human smooth muscle cells, which might promote CVD (24).
Cerebrovascular attacks (CVAs or stroke) are a leading cause of mortality and morbidity in the developed world, consisting of ischemic events in more than 80% of cases (25). Atherosclerosis plays an important role in CVAs, either by producing an embolus from a ruptured plaque or by occlusion of cerebral vessels from plaque formation (25). Two single-nucleotide polymorphisms may be present in the coding region of the CX3CR1 gene, resulting in the substitution of valine by isoleucine (V249I) and threonine by methionine (T280M) (26). The 280M allele has been found to decrease the risk of carotid artery disease (CAD) by decreasing cell adhesion, whereas data on the 249I allele have been contradictory, with both negative and positive associations with CAD (25). Kimouli et al. studied these polymorphisms to assess their association with CVAs. Their findings suggest that the M280 fractalkine receptor gene allele is associated with a lowered risk of CAD. The absence of 280M was associated with an almost fourfold risk for carotid atherosclerosis, which was higher than the risk individually conferred by conventional vascular risk factors such as age, sex, and diabetes (25). Therapeutic regulation of fractalkine/CX3CL1 has shown some initial promise. A recent study by Jiang et al. showed that aspirin inhibits TNF-α induced fractalkine/CX3CL1 expression in human umbilical vein endothelial cells (HUVECs) through the regulation of the NF-κB p65 pathway (27). Further studies are warranted to evaluate the role and molecular mechanisms regulating fractalkine/CX3CL1 in vascular pathologies.
RA is a chronic symmetric polyarticular joint disease that primarily affects the small joints of the hands and feet (9). Inflammation is characterized by infiltration of inflammatory cells such as macrophages, B cells, T cells, and dendritic cells into the joint, leading to the proliferation of synoviocytes and the destruction of cartilage and bone (1). Migration of leukocytes into the synovium is regulated through a multistep process, involving interactions between leukocytes and endothelial cells, cellular adhesion molecules, and chemokines and their receptors (1). RA pathogenesis is characterized by the recruitment and the retention of monocytes and T lymphocytes into joints (9, 28). Produced in response to inflammatory stimuli such as 1L-1β, TNF-α, IFN-γ, and lipopolysaccharide (LPS), chemokines further stimulate synovial fibroblasts and chondrocytes to release inflammatory mediators and matrix metalloproteinases, leading to cartilage degradation and pannus (i.e., newly vascularized tissue found between the bone and synovium) formation (1). Angiogenesis and cell proliferation are enhanced by chemokines, leading to synovial hyper-plasia. Synovial fibroblasts, chondrocytes, and leukocytes release chemokines that can induce autocrine and paracrine stimulation of synovial cells, resulting in tissue destruction in RA joints (1).
Fractalkine/CX3CL1 plays a dual role in RA: 1) acting as a chemotactic agent for monocytes and lymphocytes, and 2) as a cellular adhesion molecule (9). A high percentage of macrophages within RA synovium express the CX3CR1 receptor, suggesting fractalkine/CX3CL1 participates in the development of RA. Ruth et al. reported high levels of soluble fractalkine/CX3CL1 in RA SF (14). In peripheral blood and SF, mainly monocytes express frac-talkine/CX3CL1, while macrophages, fibroblasts, endothelial, and dendritic cells expressed this chemokine in synovial tissue. This study also found that a significant portion (~80%) of monocytes have CX3CR1, whereby fractalkine/CX3CL1 may recruit them to the synovium to become the primary driving force of inflammation. These levels of fractalkine/CX3CL1 in RA SF were significantly elevated compared to that in SF from osteoarthritis (OA) patients (14). Further studies have evaluated the levels of fractalkine/CX3CL1 expressing leukocytes in RA. Nanki et al. found that a substantial portion (53.4% vs. 19.2%) of RA CD8+ T cells bear surface CX3CR1 when compared to the cells derived from a healthy population (29). Administration of fractalkine/CX3CL1-specific monoclonal antibody to mice significantly reduced the clinical measure of arthritis and inflammatory infiltrate on histological sections. There was also a marked reduction in the incidence of arthritis and a reduction in the trafficking of monocytes to the synovium (29). Volin et al. demonstrated that SF depleted of fractalkine/CX3CL1 has lower angiogenic effect compared to control SF (30).
Clinical studies have shown that TNF-α inhibitors decrease fractalkine/CX3CL1 expression in RA (31, 32). Several trials have provided evidence that infliximab is safe and effective in treating RA, although some patients (20–40%) have little to no clinical response to TNF-α therapy (31). Odai et al. observed that infliximab treatment in RA inhibits the downstream signaling cascade, effectively decreasing production of inflammatory cytokines, including fractalkine/CX3CL1, in RA (32).
Following infection of cells with HIV-1, the innate immune system (specifically dendritic cells) is activated at the portal of entry. The virus is later presented to and polarizes CD4+ Th1 and Th2 cells, with the latter cells becoming infected by HIV-1 (33). CD4+ Th2 cells become depleted but harbor large amounts of virus in infected individuals (33). Mast cells, basophils, and monocytes release large amounts of IL-4, IL-5, IL-10, and IL-13, which alter the Th1/Th2 balance toward Th2 differentiation. IL-4 inhibits polarized Th1 cells from releasing IL-1, IL-2, IL-12, and IFN-γ, which are necessary for the activation of CD8+ cytotoxic lymphocyte (CTL) precursors. IL-4 also inhibits synthesis of immunoglobulin G (IgG) and IgA by B cells and induces these cells to switch to IgE synthesis. Under these conditions, the immune system is compromised and adaptive immunity is gradually inhibited (33).
Given the nature of HIV, it is not surprising that increased expression of fractalkine/CX3CL1 has been found in the lymph nodes and brain tissue of HIV patients (34, 35). The studies also found that the coadministration of fractalkine/CX3CL1 with HIV-1-induced platelet activating factor (PAF) (which acts as a neurotoxin in this context) or with the regulatory HIV-1 gene product Tat affords neuroprotection and induces the depletion of CX3CR1+ Th cells by managing contact with dendritic cells (34, 35). Fractalkine/CX3CL1 blocks the fusion of CX3CR1 with the CD4 co-receptor found on T cells, thereby potently and specifically blocking HIV-1 entry into Th2 cells (36). Polymorphisms of CX3CR1 also increase the risk for AIDS in HIV-infected patients. Faure et al. have reported that HIV-1 infected individuals homozygous for M280 progressed to AIDS more rapidly than other genotypes (37). In addition, Moatti et al. showed that HIV-1–infected patients homozygous for CX3CR1-I249M280 progressed to AIDS more rapidly than those with other haplotypes (38). These findings warrant further study to discover the role of fractalkine/CX3CL1 in the predisposition of HIV patients to develop AIDS and mechanistic approach to develop new HIV/AIDS therapies. A possible therapeutic approach using fractalkine/CX3CL1 antagonists with IL-4 antagonists in treating HIV-1 patients has been proposed (33).
Colorectal cancer is the second leading cause of cancer deaths in Western countries. Approximately one half of all patients develop metastases, especially in the liver and lung, which are the major cause of death (39). Surprisingly, fractalkine/CX3CL1 has been found to have tumor suppressive activity in various models of subcutaneous implantation in mice (39). A study by Vitale et al. showed that native fractalkine/CX3CL1 exhibits a strong antitumor effect by reducing tumor size 93% in the skin as well as 99% in orthopedic models (lung and liver) (39). Ohta et al. have reported high fractalkine/CX3CL1 expression to be a marker for better prognosis in colorectal cancer patients (40). In a recent study by Kostadinova et al., fractalkine/CX3CL1 was found to regulate the expression of inducible NO synthase (iNOS), a crucial mediator of dextran sulfate sodium (DSS)-induced colitis (41). These studies show that fractalkine/CX3CL1 plays a role in inflammatory responses but can also be beneficial with regards to tumor suppression in the viscera. However, a recent review by Marchesi et al. characterized CX3CR1 expression on a variety of tumor cells and suggested a role for CX3CR1 in aiding metastasis (42). A study by Moon et al. found suppression of TNF-α-induced fractalkine/CX3CL1 expression in endothelial cells by resveratrol, a polyphenolic compound present in grapes and red wine with anti-inflammatory, antioxidant, and antitumor properties (43). Therefore, it is clear that more studies are needed to further understand the pathophysiology of fractalkine/CX3CL1 and to provide a new method for the treatment of inflammatory bowel diseases as well as colorectal and other cancers (Figure 3).
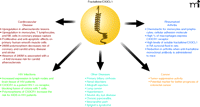
Fractalkine/CX3CL1 pathology. A summary of fractalkine/CX3CL1 involvement in complications and diseases.
Many studies have found that fractalkine/CX3CR1 participates in several other diseases. Fractalkine/CX3CL1 is expressed on endothelial cells of small vessels in normal livers and has also been detected on some (primarily damaged) bile ducts (44). Involvement in primary biliary cirrhosis has recently been discovered with lymphocyte recruitment to the liver (45). The detection of fractalkine/CX3CL1 on biliary endothelial cells in primary biliary cirrhosis and its ability to act as a chemoattractant for CD4+ T cells suggest an important role for fractalkine/CX3CL1 in the recruitment of these cells to the bile ducts (46, 47).
Fractalkine/CX3CL1 is also implicated in renal disorders, including glomerular inflammation and endothelial injury (48, 49). The expression of fractalkine/CX3CL1 is increased in renal tubulointerstitial inflammation, most notably in acute allograft rejection (48, 50). Tight junctions degenerate, possibly allowing leukocytes to escape into the urinary space in tubulointerstitial renal inflammation. Apical fractalkine/CX3CL1 may facilitate recruitment and retention of these leukocytes, redirecting them to the site of injury, where it may play a role in the development of further complications (50).
Allograft rejection is characterized by an intense cellular immune response with an influx of circulating leukocytes into the transplanted organ (51). Enhanced expression of fractalkine/CX3CL1 occurs in rejections of cardiac allografts, but treatment with CX3CR1-specific blocking antibodies significantly prolongs allograft survival (52). Other diseases in which fractalkine/CX3CL1 and CX3CR1 are involved include lung cancer, hypertension, severe dry eye disease, chronic pancreatitis, neuropathic pain, and Sjögren’s syndrome (described in Figure 3). The involvement of fractalkine/CX3CL1 in so many diseases warrants additional studies into understanding this chemokine and the role it plays in pathophysiology.
Currently there are no treatments specific for fractalkine/CX3CR1 because of the inadequate understanding of the chemokine. Several research groups have speculated ways to suppress or inhibit fractalkine/CX3CR1 expression. Elucidation of the structure of fractalkine/CX3CL1 has made it possible to develop synthetic or biological analogs to inhibit CX3CR1. Inoue et al. developed frac-talkine/CX3CL1 analogs by truncating four N-terminal amino acids from native fractalkine/CX3CL1. The effect of these analogs was to inhibit fractalkine/CX3CL1-mediated chemotaxis in CX3CR1-expressing glomerular endothelial cells (53).
Drugs currently available are being studied to determine their abilities to target fractalkine/CX3CL1. Recently, Duthey et al. tested receptor agonist/the anti-inflammatory effects of baclofen (GABAB antispastic agent) on allergic contact dermatitis. The study found baclofen induces heterologous desensitization of several chemokine receptors including CX3CR1 (54). Lee et al. found that epigallocat-echin-3-O-gallate (EGCG) decreases TNF-α induced fractalkine/CX3CL1 expression in HUVECs through NF-κB pathway regulation (55). Wan et al. investigated the effects of rosiglitazone (PPARγ agonist/insulin sensitizer) on fractalkine/CX3CL1 expression (56). The activation of PPARγ by rosiglitazone in macrophages repressed the transcription of the fractalkine/CX3CL1 gene and prevented plasma membrane translocation. The activation of PPARγ by rosiglitazone in endothelial cells also impeded the nuclear export of fractalkine/CX3CL1, suggesting that PPARγ activation may suppress fractalkine/CX3CL1 signaling (56).
In summary, fractalkine/CX3CL1 is a unique chemokine made of 373 total amino acids, consisting of an extracellular N terminal domain, a mucin-like stalk, a transmembrane α helix, and a short cytoplasmic tail. The soluble form of fractalkine/CX3CL1 exhibits chemotactic activity for monocytes, NK cells, and T cells. Also, fractalkine/CX3CL1 acts as an adhesion molecule to leukocytes (16) and enhances extravasation of leukocytes through the endothelia (10). Induced by TNF-α, IFN-γ, and IL-1β, fractalkine/CX3CL1 is actively involved in many complications and diseases such as atherosclerosis, RA, HIV infection, cancer, and several other disorders. Although no specific clinical treatment is currently available to target fractalkine/CX3CL1, common drugs and new antagonists are being tested to develop new therapeutic approaches to diseases. Researching this novel chemokine for a better understanding of its role in pathogenesis may lead to new therapies directed toward fractalkine/CX3CL1 regulation.
Acknowledgments
This work was supported in part by the National Institutes of Health [Grants AT-003633, AR-055741] and by the start-up funds from The University of Toledo. The authors thank Ms. Charisse N. Montgomery for critical reading of the review.
- Copyright © 2010
References

Salahuddin Ahmed, PhD, is an Assistant Professor of Pharmacology in the College of Pharmacy at the University of Toledo, OH. His research group is involved in studying the molecular mechanisms that drive unwanted extracellular matrix remodeling and joint destruction in rheumatoid arthritis (RA) and osteoarthritis (OA). His group has pioneered in defining the mechanisms through which polyphenol in green tea, epigallocatechin-3-gallate (EGCG), regulates chemokine and proinflammatory cytokine mediated signaling events that result in joint destruction in RA and OA. E-mail salah.ahmed{at}utoledo.edu; fax 419-383-1909.

Brian A. Jones is an undergraduate student in the Ahmed laboratory in the College of Pharmacy at the University of Toledo. This review was written as part of his summer internship and has aided in his understanding of the role of fractalkine/CX3CL1 in the development of rheumatoid arthritis. The primary research interests that he plans to pursue include developing novel therapeutic entities against autoimmune diseases and drug design and discovery.

Maria Beamer, BS, is a Research Assistant in the Ahmed laboratory in the College of Pharmacy at the University of Toledo. She graduated in biochemistry/cell biology from the University of California at San Diego. She is involved in studies related to the regulation of angiogenesis and joint destruction by green tea polyphenol epigallo-catechin-3-gallate (EGCG) in rheumatoid arthritis.

