Spindle Poisons and Cell Fate: A Tale of Two Pathways
Abstract
Spindle poisons, such as paclitaxel and vinblastine, exert their potent anti-neoplastic effects through activation of the spindle assembly checkpoint (SAC), thereby arresting cells in mitosis. Unfortunately, only certain cancers are susceptible to these drugs, and many patients fail to respond to treatment. We review the pathways that are triggered by spindle poisons and highlight recent studies that describe the great variability of tumor cells in responding to these drugs. We also describe the recent identification of an apoptotic pathway that is activated by mitotic arrest in response to spindle poisons. Emerging from these studies is not only a greater understanding of how these classic antimitotic agents bring about cell death, but also a wealth of potential new targets of anticancer therapeutics.
Introduction
Spindle poisons are some of the oldest and most successful chemotherapeutics (1). These drugs bind tubulin, a protein that forms the central constituent of the microtubule cytoskeleton and has many cellular functions. The formation of the mitotic spindle is one of the most fundamental of cellular functions that is provided by tubulin, allowing for the equal division of chromosomes during mitosis. By interfering with the normal dynamic activity of microtubules, spindle poisons bring about a prolonged mitotic arrest. It is widely believed that this prolonged arrest is central to spindle poison–induced cell death, but the mechanistic details by which these drugs kill cells remain unclear.
Microtubules are polymers of tubulin and constitute a major component of cytoskeletal networks. Cells constantly change the lengths of their microtubules and this process is tightly controlled by properties that are intrinsic to tubulin as well as by additional proteins that regulate tubulin polymerization. The dynamic regulation of microtubule length is central to microtubule function. Paclitaxel and the vinca alkaloid vinblastine are two prototypical spindle poisons used in the treatment of ovarian, breast, and several lymphoid-derived cancers, as well as cancers of the head, neck, and lung (2, 3). Although both drugs are thought to kill cells by a mode of prolonged mitotic arrest, their effects on microtubules are distinct. Paclitaxel binds to tubulin within existing microtubules and stabilizes the polymer, whereas vinblastine targets tubulin monomers and prevents their addition to the microtubule terminus, ultimately resulting in the absence of polymerized microtubules. By interfering with microtubule dynamics, both classes of drugs preclude the normal function of the mitotic spindle and arrest cells in mitosis. Many derivatives of paclitaxel and vinblastine have been synthesized, generating numerous spindle toxins, and better analogs and improved formulations of these drugs have recently been brought to market (4, 5). We will concentrate on the pathways that are stimulated in cells exposed to spindle poisons; for discussions of individual compounds and their clinical efficacies, the reader may consult published reviews (2, 6).
Unfortunately, the development of resistance to these drugs prior to complete tumor eradication is common, resulting in significant clinical challenges. Drug resistance is often ascribed to drug efflux pumps or mutations in tubulin that abrogate drug binding, although these clearly do not account for all cases (7–9). Investigations into other mechanisms by which cells evade death by spindle poisons may thus be crucial for efforts to improve the efficacy of existing drugs and expand our search for novel therapies.
Mitosis and the Spindle Assembly Checkpoint (SAC)
During mitosis, cells replicate their genetic material and divide it equally into two daughter cells. Interfering with this tightly orchestrated distribution of genetic material can generate cells with irregular chromosome content (a condition known as aneuploidy), as well as genomic instability, cell death, and possibly cancer (10–13). Not surprisingly, cells have evolved numerous mechanisms to ensure that mitosis is accomplished with great fidelity.
Early investigators found that when cells were incubated with spindle poisons the number of cells in mitosis dramatically increased over time as cells entered mitosis but failed to exit. Elegant research over the last twenty years identified the spindle assembly checkpoint (SAC) as the signaling pathway responsible for this mitotic arrest. This checkpoint evolved to ensure accurate segregation of chromosomes. The signal emerges from kinetochores, which form the protein link between microtubules and DNA on each chromatid (14–16). A single unattached kinetochore can generate enough signal to block cell cycle progression, ensuring that every chromosome is correctly attached to the spindle apparatus prior to anaphase (17–20).
The activation of cyclin-dependent kinase 1 (CDK1) by cyclin B drives a cell into mitosis, at which point a complex called cohesin holds the sister chromatids together in a mitotic chromosome. When the mitotic cell progresses to metaphase, the SAC becomes responsible for controlling exit from mitosis (Figure 1). In a normal mitosis, the SAC is active only briefly (a period of minutes) as unattached chromosomes attach to spindle microtubules and correct improper attachments. During this time, SAC activity maintains high cyclin B levels through inhibition of an E3 ubiquitin ligase that normally targets cyclin B for degradation (Figure 1A). This ubiquitin ligase is known as the anaphase promoting complex or cyclosome (APC/C). By inhibiting the APC/C, the SAC keeps cyclin B levels high and ultimately protects CDK1 and cohesin activity. When all chromosomes are properly attached, the SAC is turned off and the APC/C degrades cyclin B. This drop in cyclin B levels results in exit from mitosis, and both CDK1 and a protein called securin (which protects cohesin) are targeted (also by the APC/C) for degradation (Figure 1B) (21).
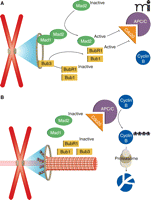
By inhibiting the degradation of cyclin B by the anaphase promoting complex/cyclosome (APC/C), the SAC arrests the cell in metaphase until all kinetochores are attached to spindle microtubules. A. In the absence of microtubule attachments, unattached kinetochores recruit checkpoint proteins and activate them, allowing Mad2, Bub3, and BubR1 to inhibit Cdc20 activation of APC/C, resulting in high amounts cyclin B and metaphase arrest. B. When all kinetochores have proper microtubule attachments, checkpoint proteins are no longer recruited to kinetochores and Cdc20 activates APC/C, which drives cyclin B degradation. Depletion of cyclin B causes anaphase onset.
In contrast, when cells enter mitosis in the presence of spindle poisons, kinetochores are never able to form proper attachments to spindle microtubules. This results in the permanent activation of the SAC and a mitotic arrest that lasts for hours. These cells will eventually either apoptose or exit mitosis by an alternate mechanism that does not occur during a normal mitosis and which we will discuss below. Thus, the SAC is briefly active during a typical mitosis until all kinetochores are attached to spindle microtubules. The SAC also frequently responds to attachment defects that occur under normal conditions by arresting cells in mitosis until errors are resolved. However, spindle poisons greatly extend the duration of this arrest and prevent cells from exiting mitosis correctly.
The SAC inhibits the APC/C by preventing its activation. The ubiquitin ligase activity of the APC/C requires activation by Cdc20 (22). When the checkpoint is active, two SAC proteins called Mad2 and BubR1 (assisted by Bub1) prevent APC/C activation by binding the Cdc20-APC/C complex. When all kinetochores have proper attachments and the SAC is inactive, Cdc20 is free to activate the APC/C, quickly leading to cyclin B degradation and anaphase onset (Figure 1B). Importantly, even during a robust SAC response, cyclin B degradation still takes place slowly owing to incomplete inhibition of the APC/C. This phenomenon, known as “slippage”, means that cells treated with spindle poisons can eventually exit mitosis despite an active SAC (23).
The propagation of the SAC signal from a single unattached kinetochore to Cdc20-APC/C complexes located throughout the cell is accomplished via two groups of checkpoint proteins originally identified in independent genetic screens. Mitotic arrest deficient (Mad1, Mad2, and Mad3/BubR1) and budding uninhibited by benzimidazoles (Bub1) genes were found to be required for Schizosaccharomyces pombe to arrest normally in response to spindle poisons (24, 25). Further study revealed that these highly conserved proteins (later to include Bub3 in the pathway) are recruited to improperly attached kinetochores, at which time they are activated and released to bind and inhibit Cdc20-APC/C complexes. Although Mad1 and Mad2 seem to be activated independently of Bub1, Bub3, and BubR1, the two groups ultimately cooperate to inhibit APC/C activation.
The mechanism of spindle checkpoint signaling is still an area of active research and it is likely that additional proteins, and thus additional new targets, will be identified. While the Mad and Bub proteins remain the major bona fide checkpoint proteins within the SAC, they have been joined by a number of secondary players that also function in this pathway (26–29). It is still unclear how kinetochores generate the signal, but recently over 60 new kinetochore proteins have been identified which should facilitate the identification of this important activity (30, 31).
Variations in the Cellular Response to Checkpoint Activation
Given the long clinical history of drugs like paclitaxel and vinblastine and the immense degree of study that has been put forth to understand the mechanisms of action of these and similar drugs, it is surprising how little is understood about how these compounds promote cell death (32). As these compounds lead to permanent activation of the SAC, it is widely assumed that they kill cancer cells through a mechanism of mitotic arrest, but this idea has been challenged repeatedly (33, 34). It is clear that many compounds that result in mitotic arrest are neither useful therapeutics nor even effective at killing cells. While this may arise from bioavailability or dosage issues, it suggests that artificially prolonging the time cells spend in mitosis is not in itself sufficient to kill cells. Moreover, some cancers are incredibly sensitive to treatment with paclitaxel, whereas others are virtually non-responsive (35). This variation presents a serious clinical problem that has defied the development of predictive markers and genetic screens (36).
In an effort to better understand the cellular response to spindle poisons, investigators typically have turned to studying responses in vitro. For instance, assaying the percentage of apoptotic cells following drug treatment allows for comparison between cell lines to determine which is more sensitive to the drug. Although these studies are simple to perform, they fail to capture the variety of responses that may occur within a population of cells. For some time, we have assumed that all cells that die within a culture that is exposed to paclitaxel, for example, represent a homogeneous fate. Likewise, cells that survive have also been assumed to reflect a single mode of response to drug treatment. Recent studies, however, have utilized high-throughput single-cell imaging to demonstrate that remarkable variation exists among like cells responding to antimitotic agents (37, 38). Automated microscopy systems that image cells over prolonged periods allow researchers to follow the fate of single cells treated with a drug. Cells expressing a fluorescent chromatin marker (such as red fluorescent protein–tagged histone H2B) are readily monitored over time. Videos of the cells allow investigators to analyze and score cell fates into categories: (e.g., mitotic exit, cell division, death in mitosis, death in interphase, no mitotic entry, etc.). The behavior and fates of hundreds of individual cells can, in this way, be studied.
Upon treatment with the microtubule-depolymerizing drug nocodazole, for example, the majority (63%) of HeLa cells survive, either in interphase following mitosis (8%) or after exiting mitosis and duplicating their genome repeatedly without subsequent cell divisions, a process known as endocycling (55%) (Figure 2) (37). The cells that die can be divided into five categories, and only 6% of the total exhibit the cell fate most commonly associated with spindle poisons, namely, death in mitosis after a prolonged arrest. Significantly, had the response of these cells been examined by simply counting the number of apoptotic cells at the conclusion of the experiment, the investigators could have only concluded that 37% of the cells had died. The reality is that these 37% of nocodazole treated cells display five unique fates following treatment and only a minority dies by the presumed mode ascribed to spindle poisons (Figure 2).
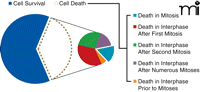
Population level analyses reveal the percentage of HeLa cells that survive (blue) or apoptose (white) following treatment with nocodazole. Analysis of individual cells reveals the large amount of variation in how the cells die (smaller pie). Only a small minority of HeLa cells treated with nocodazole die during the first mitotic arrest, which is the presumed mode by which spindle toxins bring about cell death. Cells that survive represent both survival in interphase after mitosis (8%) and survival after replicating their genomes but failing to complete mitosis, i.e. endocycling (55%).
Strikingly, the same HeLa cells treated with a different spindle poison, paclitaxel, exhibited a widely different set of fates, despite the fact that paclitaxel similarly arrests cells in mitosis. The majority of paclitaxel treated cells (73%) exited mitosis after a mitotic arrest and then died in the subsequent interphase. Differing degrees of this within-group variance were seen across fifteen different cell lines. This within-group variance does not arise from genetic differences within individual cell lines, as clonally expanded cells exhibit the same set of fates as the parental line (37). This result is particularly perplexing. How is it that genetically identical cells (expanded from a single clone) can respond to mitotic arrest in fundamentally different ways?
In an effort to address this question, the authors inhibited caspase activation during mitotic arrest and observed that it completely prevented cell death (37). By inhibiting the induction of apoptosis, cells that would have died instead exited into interphase. This suggested that apoptosis and mitotic exit are two possible fates following mitotic arrest. Although the spindle assembly checkpoint pathway is responsible for controlling mitotic exit, it does not play any direct role in the induction of apoptosis. This led to the hypothesis of a separate pathway that controlled the induction of apoptosis during mitotic arrest.
A Pro-Apoptotic Pathway in Mitosis
Apoptosis, or programmed cell death, occurs in response to a number of intracellular and extracellular stimuli (39) and can be triggered by several pathways that monitor DNA damage, nuclear instability, and oxidative stress. These distinct pathways converge on the activation of caspases and other terminal events, including membrane blebbing and fragmentation of genomic DNA. Mutations that suppress apoptotic pathways can also be found in oncogenes, suggesting that apoptosis is tied into a broader network that monitors the cellular state for cancerous traits (40). Interestingly, previous studies also linked the apoptotic network to mitotic duration and suggested that maximizing the time that cells spend in mitosis could be a goal of future therapies (41). However, until recently a mechanism through which the cell would actually monitor the duration of mitosis and link this to the activation of cell death pathways has not been reported. New studies have identified a pathway involving the protein Mcl1 that couples the timing of mitosis to the induction of apoptosis (42). The results implicate Mcl1 in a newly-discovered apoptotic pathway that is active in mitosis (Figure 3).
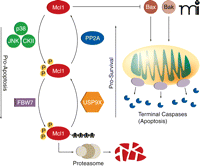
During prolonged mitosis, Mcl1 prevents the induction of apoptosis by inhibiting the binding of Bak and Bax to the mitochondrial membrane. However, Mcl1 levels fall over time. The protein kinases p38, Jun N-terminal kinase (JNK), and casein kinase II (CKII) phosphorylate Mcl1, driving its interaction with FBW7, the substrate binding component of a ubiquitin ligase complex. Ubiquitinated Mcl1 is then degraded by the proteasome. The deubiquitinase USP9X and protein phosphatase PP2A can promote Mcl1 stability by removing ubiquitin side-chains and dephosphorylating Mcl1, respectively. When Mcl1 concentrations fall low enough, Bak and Bax form pores in the mitochondrial membrane, resulting in the release of cytochrome c and terminal caspase activation and in apoptosis.
Mcl1 is a unique member of the Bcl2 family of pro-survival proteins (43, 44). Similar to other Bcl2 family members, Mcl1 binds to Bak and Bax and inhibits their association with the mitochondrial membrane. In this way, Mcl1 prevents Bak and Bax from forming pores in the mitochondrial membrane and initiating apoptosis. Unlike Bcl2, whose expression remains fairly constant and only decreases when cells approach terminal differentiation, Mcl1 expression appears to be temporarily increased at critical periods, including mitotic arrest during treatment with spindle poisons (45). This enables the cell to better inhibit the apoptotic pathway in the face of powerful insults which would otherwise cause the immediate induction of cell death. Perhaps the purpose of this is to buy the cell time as it struggles to correct errors or perform important cell fate decisions. However, because the Mcl1 elevation is transient, apoptosis is eventually initiated if the cell does not correct the offending problem during this period of high Mcl1 (42).
When Mcl1 expression is induced following mitotic arrest, its degradation is carefully regulated by several important factors (Figure 3) (42). FBW7, the substrate-binding component of a ubiquitin ligase complex, targets Mcl1 for degradation by the 26S proteasome, whereas the deubiquitinase USP9X counteracts the polyubiquitinating activity of the FBW7 complex (46). During mitotic arrest, the activities of Jun N-terminal kinase (JNK), the mitogen-activated protein kinase (MAPK) family member p38, and casein kinase II (CKII) are increased, leading to phosphorylation of Mcl1, interaction with FBW7, and the subsequent polyubiquitination of Mcl1. Polyubiquitylation of Mcl1 leads to degradation via the proteasome and continually decreasing concentrations of Mcl1 during mitotic arrest. If the arrest is alleviated, then JNK, p38, and CKII activity quickly falls and PP2A rapidly dephosphorylates Mcl1, abrogating its interaction with FBW7 and keeping Mcl1 amounts in the cell high.
The peak levels of Mcl1 occur immediately following the addition of spindle poisons (at the onset of mitotic arrest), providing time for the cell to resolve errors in kinetochore-microtubule attachment (Figure 4). During this period, the amount of Mcl1 slowly decreases. Concurrently, although the SAC is active cyclin B amounts still slowly decline because of slippage mediated degradation by the APC/C. If the SAC can be silenced during this period via the attachment of kinetochores to spindle microtubules, Mcl1 levels will be stabilized and cyclin B will be degraded, leading to mitotic exit. However, if (in the case of prolonged treatment with spindle poisons) kinetochores never correctly attach to spindle microtubules and the SAC remains active, the rate at which Mcl1 and cyclin B are degraded ultimately determines the fate of the cell. If cyclin B levels first fall below the threshold for mitotic exit, then the cell will enter anaphase before cell death is initiated (Figure 4B). However, if Mcl1 is degraded past the levels necessary to inhibit Bak and Bax, the apoptotic pathway will be triggered before the cell can exit mitosis (Figure 4C).
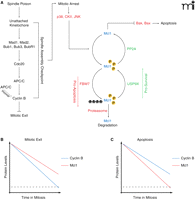
A. SAC activation leads to mitotic arrest and triggers the onset of Mcl1 degradation. Mcl1 is acted on by a set of pro-apoptotic proteins that drive its degradation and a set of pro-survival proteins that enhance its stability. During prolonged mitotic arrest, the pro-apoptotic pathway is favored. Simultaneously, incomplete inhibition of the APC/C results in slow cyclin B degradation, a phenomenon known as slippage. B. Cells treated with spindle poisons can exit mitosis if cyclin B concentrations fall below the threshold (dotted line) before the concentration of Mcl1 does. C. If Mcl1 levels fall below threshold (dotted line) before cyclin B, the apoptotic pathway is triggered while the cell is in mitosis.
Supporting these findings, studies of cancer cell lines and patient-derived tumor samples reveal the presence of elevated Mcl1 expression (47–49). Closer examination has also revealed a correlation between Mcl1 amounts and progression to malignancy within individual cancers (48). It is not yet completely clear what leads to the increased amount of Mcl1, although increased activity of the Mcl1 deubuitinase USP9X has been correlated with poor prognosis in patients with multiple myeloma.
Slippage as a Target of Anti-Mitotic Drugs
Given our understanding of the control of apoptosis during mitotic arrest, two approaches to increasing the efficacy of spindle poisons are: 1) prolonging the duration of mitotic arrest and 2) enhancing the degradation of the apoptotic timer. Stated differently, in order to favor apoptosis over mitotic exit, the rate of cyclin B degradation must be slowed or the rate of Mcl1 degradation must be increased. The end goal of either approach is to ensure that Mcl1 levels breach the apoptotic threshold before cyclin B levels fall below that required for mitotic exit. In fact, it might be ideal to identify drugs that work on both pathways, as one would expect their activities to be synergistic.
Many drugs, in addition to the spindle poisons, have been developed that stabilize cyclin B concentrations. The same pathway of SAC activation and APC/C inhibition that paclitaxel and vinblastine activate has been fairly well-elucidated. The challenge in this case is that even in the complete absence of microtubules, when the SAC should be maximally activated, slippage or weak degradation of cyclin B still occurs (23) despite the fact that cyclin B is degraded only by the activity of the APC/C. It seems unlikely that attempting to augment the activity of the SAC upstream of the APC/C will prevent the degradation of cyclin B characterized by slippage. With slippage as the target, inhibition of either the APC/C or the 26S proteasome represent two possible options.
Knock-down of the APC/C by RNA interference leads to mitotic arrest, and small-molecules have been identified and characterized that inhibit APC/C activity, suggesting that this E3 ubiquitin ligase is a worthy target for future research and, possibly, therapy (50, 51). As well, APC/C inhibition, as part of a combination drug regimen, may sensitize cells to existing chemotherapeutics; however, the design of drugs that target the APC/C is hampered by the fact that ubiquitin ligases are traditionally difficult targets for small molecules.
The 26S proteasome is a considerably better-studied target. This proteasome is responsible for degrading cyclin B following its polyubiquitination by the APC/C. However, because this complex is also important for the degradation of Mcl1, the pharmacological manipulation of the proteasome might be complicated. Nonetheless, subunits of the proteasome were identified in at least one large RNA interference screen designed to identify genes that sensitize non-small-cell lung cancer cells to paclitaxel (52). MG132 is one small-molecule inhibitor of the proteasome whose effects have been studied extensively (53–55). Treating cells with MG132 stabilizes cyclin B levels and arrests cells in mitosis for prolonged periods. The inhibitor also increases the apoptotic effects of paclitaxel in a number of cancer cell lines, although it is not clear whether these effects arise from a decreased rate of cyclin B degradation over that observed with paclitaxel treatment alone (56, 57). Although MG132 was originally designed primarily to aid laboratory study of the proteasome, a related small molecule (PS431, Bortezomib) was subsequently developed for cancer therapy (58, 59). Despite the fact that the proteasome participates in a number of diverse cellular processes, the side effects of Bortezomib treatment were surprisingly mild and the drug has received FDA approval. It is currently a second-line therapy for multiple myeloma and relapsed mantle cell lymphoma. Intriguingly, co-treatment of Bortezomib with docetaxel (a paclitaxel analog) greatly enhances apoptosis in several gastric cancer cell lines over apoptosis observed with treatment by either drug alone; these results were mirrored in another study examining head and neck cancer lines (60). Combination studies of Bortezomib with paclitaxel or docetaxel are ongoing and have yet to enter Phase III studies, although reported side effects are similar to treatment with Bortezomib alone (61).
Enhancing MCL1 Degradation to Promote Apoptosis
The three protein serine-threonine kinases that directly phosphorylate Mcl1 and favor its interaction with FBW7 are JNK, p38, and CKII (46, 62, 63). To promote Mcl1 degradation, one might try to maximize the activity of these enzymes, but it isn’t clear whether a small molecule could be rationally designed to do this. Nevertheless, JNK and p38 have been studied extensively (64–67), and small-molecule inhibitors of JNK are in clinical trials that target diseases such as cancer, whereas inhibitors of p38 have undergone clinical trials for the treatment of cancer and rheumatoid arthritis (68, 69). Both of these protein kinases regulate stress responses and inflammation but have also been linked to apoptosis. Indeed, both pro-apoptotic and anti-apoptotic roles have been ascribed to these proteins (70), and their functions in apoptosis may vary by cell line and be dependent on cell cycle stage and other cellular conditions. Although inhibition of JNK and p38 sensitizes certain cell lines to treatment with chemotherapeutics, it isn’t clear whether this effect is mediated via degradation of Mcl1 or through alternate pathways. Similarly, CKII participates in a complex number of cellular functions and has anti-apoptotic effects (71). Inhibition of CKII can also trigger apoptosis and sensitive cells to antimitotics (72).
These somewhat paradoxical results are difficult to analyze. Historically, the functions of JNK, p38, and CKII have often been linked to cell survival. Their myriad roles in proliferation, differentiation, and (especially) the stress response, would seem to confound studies seeking to identify any direct role in apoptosis. It is conceivable that treatment with spindle poisons would activate the stress response pathways mediated by these kinases. Their increased activity would then also lead to Mcl1 phosphorylation, driving Mcl1 degradation. It would make sense to test whether further stressing cells to activate the stress response pathway could increase the percentage of cells that die in mitosis. This hypothesis is supported by the results of unbiased screens that knocked-down the expression of all human protein kinases to identify proteins whose absence increases the sensitivity of cells to paclitaxel. In this study, the gene COL4A3BP, which encodes a protein serine-threonine kinase, caused the most striking sensitivity (73). Loss of this protein, which is involved in ceramide metabolism, can activate the ER unfolding stress response pathway, which might activate stress kinases (e.g., JNK and p38). It will be important to determine whether the spindle poisons whose use leads to cell death also activate the stress response more than those drugs that simply inhibit mitotic exit.
A more rational target is the protein phosphatase PP2A, which is believed to dephosphorylate Mcl1 and thereby abrogate Mcl1’s interaction with FBW7. PP2A is an attractive target because inhibiting the effects of this phosphatase should be equivalent to increasing the activities of JNK, p38, and CKII (Figure 3). Moreover, the association of PP2A with Mcl1 is already decreased during prolonged mitotic arrest, likely contributing to the ability of JNK, p38, and CKII to phosphorylate Mcl1. There also are already well-established PP2A inhibitors, such as okadaic acid (74). Treating cells with okadaic acid leads to results that are similar to those seen after paclitaxel treatment, such as defective spindle formation and mitotic arrest. Moreover, treating cells with okadaic acid during mitosis leads to increased phosphorylation of Bcl2, which also promotes apoptosis (75). Thus, inhibiting the function of PP2A may favor the induction of apoptosis by acting indirectly through multiple targets. Inhibition of PP2A could be complicated, however, as it is used in many cellular processes and has also been implicated in APC/C activation.
The ubiquitination of Mcl1 may represent another possible target for molecular intervention to promote apotosis. Inhibition of the deubiquitinase USP9X, which counteracts FBW7 by removing ubiquitin side chains from Mcl1, would promote increased degradation of Mcl1 by the proteasome. Knock-down of USP9X results in decreased levels of Mcl1 protein and sensitizes cells to treatment with paclitaxel (46). Importantly, the catalytic site of USP9X has been identified. USP9X has also been implicated in tumor growth factor β signaling, tight-junction assembly, and other processes outside of mitosis, but the off-target effects of any putative USP9X inhibitor aren’t clear, as no known inhibitor exists (76, 77). Still, USP9X may represent the best target within the Mcl1 pathway.
Conclusions
Decades of work on the SAC have complemented recent studies to paint a fascinating portrait of how this cellular process promotes cell death. Improving the efficacy of drugs like paclitaxel and vinblastine can be rationally approached from two angles. Helping spindle poisons to better stabilize cyclin B by reducing the effects of slippage would allow the checkpoint sufficient time for Mcl1 concentrations to fall below the apoptotic threshold. On the other hand, increasing the rate of Mcl1 degradation will allow the induction of apoptosis to occur earlier. Targeting these pathways together will ideally yield an approach that both arrests cells in a prolonged mitosis and steers them toward an apoptotic fate.
Alongside any drug development, a great deal of work remains to be done to further elucidate the connections between the SAC, Mcl1, and cell fate. For example, slippage is APC/C dependent, but it isn’t clear why this complex is incompletely inhibited by the SAC. The existence of alternate pathways that act on the APC/C to modulate its activity should be explored as possible sources of the slippage mechanism. The numerous phosphorylation sites on APC/C could represent a starting point for future studies (78).
Another issue is why different spindle poison drugs, which are all believed to act via a common mechanism of prolonged SAC activation, nonetheless result in highly dissimilar cellular fates. Is this because drugs such as paclitaxel and vinblastine activate the SAC in dissimilar ways or is it possibly because of dissimilar activities of these drugs on stress pathways? Similarly, it is unclear why taxanes and vinca alkaloids are effective chemotherapeutics whereas microtubule depolymerizing drugs like nocodazole are not effective. Because all of these drugs activate the spindle checkpoint, the important difference between them may reside in other pathways that are activated during treatment. Notably, the most potent spindle poisons bind non-reversibly whereas nocodazole can be easily washed out of cells. Non-reversible binding may be required in order to maintain effective doses in patients long enough for these drugs to work. Cells are only in mitosis for a short period of the cell division cycle, thus only a small percentage of cells may traverse mitosis during the period that a reversible drug is present at adequate intracellular concentrations. This issue of reversible and non-reversible binding will be an important consideration in the design of new anti-mitotic drugs with adequate bioavailability.
Stress kinases such as JNK, p38, and CKII may participate in promoting apoptosis by phosphorylating Mcl1. It’s possible that indiscriminate activation of the stress response pathway may sensitize cells to killing by spindle poisons and perhaps by other chemotherapeutics as well. This idea could explain why some drugs seem to have additive effects when administered together, despite the fact that they don’t act on common pathways. In fact, one drug may simply be activating the stress response pathway and making the cell more generally susceptible to treatment with another.
Finally, innovative studies need to explore the diverse factors that sensitize or protect cells from the effects of paclitaxel and related drugs. For example, two RNA interference screens performed in human cells identified genes responsible for mitotic regulation as being important for the cellular response to paclitaxel (52, 73). However, these studies also identified dozens of other genes as being critical to the paclitaxel response and their functions are as diverse as actin polymerization and ceramide metabolism. Determining how these diverse processes feed into the cellular response to spindle poisons is a promising area for future research.
The number of additional genes that may be ultimately responsible for the paclitaxel response remains unknown, but the recent results we have highlighted should serve as encouragement to those who believe that additional drug targets must exist within this established therapeutic pathway. These recent developments continue to highlight the importance of the SAC and apoptosis in both basic biology and disease treatment.
Acknowledgments
We would like to thank Dr. Vishva Dixit (Genentech Corporation) for advance viewing of prepublication material and Dr. John Densmore (University of Virginia) for critical review of the manuscript. This work was supported by the National Institutes of Health [Grant 5R01GM081576-03]. DRM was also supported by funding from the National Institutes of Health [Grant T32 GM008136 ] and the University of Virginia Medical Scientist Training Program [Grant T32 GM007267-33 ].
Footnotes
-
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Matson and Stukenberg.
- Copyright © 2011
References
Todd Stukenberg, PhD, is a tenured Professor of Biochemistry at the University of Virginia. His work focuses on the kinetochore and the regulation of mitosis in Xenopus laevis and tissue culture cells. E-mail pts7h{at}virginia.edu; fax 434-924-5069.

Daniel Matson is an MD/PhD candidate investigating the role of centromere components in the spindle assembly checkpoint. He is training with Todd Stukenberg in the Department of Biochemistry and Molecular Genetics at the University of Virginia.


