Emerging Roles of TACE as a Key Protease in ErbB Ligand Shedding
The epidermal growth factor (EGF) family and their receptors—the ErbB family of type I receptor tyrosine kinases—participate in development, differentiation, proliferation, and survival (1,2). Hyperactivity of ErbB signaling has also been strongly implicated in human cancer (3), whereas targeted mutation of ErbB1/EGFR in mice results in embryonic and perinatal lethality with epithelial cells being most profoundly affected by receptor loss (4–,6). All endogenous ErbB ligands are synthesized as transmembrane precursors that are proteolytically cleaved (see Table 1⇓) to release biologically active soluble growth factors, which act in an autocrine or paracrine manner (7) (Figure 1⇓). Although in vitro studies have indicated that membrane-anchored ErbB ligand precursors may also be biologically active at the cell surface in a juxtacrine manner (8–,11), genetic studies in Drosophila (12–,14) and blockade of shedding in mammalian cells (15) highlight a requirement for proteolytic release of ErbB ligands.
The regulation of ErbB ligand presentation and processing has been an area of intense investigation over the last decade and many questions have been raised. Why are ErbB ligands synthesized as transmembrane precursors? Do membrane-anchored ligand precursors and shed soluble ligands have different signaling capacities? How is the proteolytic release of ligands regulated, and what proteases are involved? Progress in regard to better defining the proteases involved in ErbB ligand release, highlighted by the recent publication of Sunnarborg et al., suggests a larger role for the tumor necrosis factor-α (TNFα) converting enzyme (TACE/ADAM17) (16).
TACE, a member of a large family of disintegrin-metalloproteinases (ADAMs), is capable of processing diverse substrates including pro-transforming growth factor-α (proTGFα), TNFα, and L-selectin (17). Mice that produce catalytically inactive TACE protein that cannot bind Zn2+ (Tace ΔZn/ΔZn) display a severe phenotype similar to the embryonic and perinatal lethality observed in EGFR-deficient mice (4–,6). The eye, hair, and skin phenotypes of newborn Tace ΔZn/ΔZn mice are identical to those previously reported for TGFα-deficient mice (18,19). Indeed, the involvement of TACE in proTGFα processing was confirmed by the reduced shedding of endogenous TGFα from ras- and myc-transformed embryonic fibroblasts derived from Tace ΔZn/ΔZn mice, and by the ability of recombinant TACE to correctly process a proTGFα juxtamembrane peptide in vitro. Mice lacking functional TACE have additional defects beyond those observed in TGFα–deficient mice, including defects in: epithelial maturation within multiple organs; formation of the lung vasculature; airway branching; and the spongiotrophoblast layer of the placenta (17,20). Both the decreased size and the delayed cellular differentiation of TACE-deficient lungs are restored by the exogenous addition of soluble EGF in embryonic lung culture (20). Together, these genetic data further support the concept that TACE-dependent cleavage of proTGFα and other ErbB ligands is essential to development.
The studies by Sunnarborg et al. provide an excellent example of the key criteria needed to unambiguously define a role for TACE in processing of proTGFα. Previous genetic studies have relied on examining the loss of substrate shedding in ras- and myc-transformed embryonic fibroblasts from Tace ΔZn/ΔZn mice, however it is unclear how transformation and constitutively active Ras signaling may affect proteolytic cleavage. In contrast, Sunnarborg et al. demonstrate defective TGFα shedding in primary cultures of normal Tace ΔZn/ΔZn keratinocytes, and partially restore shedding by re-expressing TACE in these cells.
Another major limitation in defining the participation of specific ADAMs in ErbB ligand processing has been the difficulty of demonstrating the direct cleavage of ligands, and verifying that the ligands have been processed at the correct sites of proteolysis. In the case of TACE, in vitro assays demonstrated the proteolytic cleavage of TNFα, L-selectin, and TGFα juxtamembrane peptides (17,21,22). Sunnarborg et al. have extended this approach to proTGFα and its N- and C-terminal cleavage site peptides, demonstrating that TACE can correctly cleave both N- and C-terminal cleavage sites. Interestingly, most TACE substrates are cleaved within a short stretch of amino acids located in the juxtamembrane region of the extracellular domain. The possibility that TACE may be involved in both N- and C-terminal proTGFα cleavage events raises important questions about how TACE, which is an integral transmembrane protein, can recognize and discriminate between cleavage sites that are located within the extracellular domain of the same protein.
The importance of ErbB ligand shedding has also led investigators to examine the regulation and specificity of TACE and other metalloproteinases in the processing of other ErbB ligands. Indeed, the sequential cleavage of the extracellular domain of membrane-anchored EGF-like growth factors is a highly regulated process that can be activated by a wide variety of pharmacological agents as well as physiological stimuli (23–,25). Using different experimental approaches, studies have implicated matrix metalloproteinases as well as ADAM proteases in the proteolytic cleavage and shedding of ErbB ligands (Table 1⇓). The work by Sunnarborg et al. adds two other ErbB ligands to the list of TACE substrates, with their demonstration that TACE restores cleavage of transfected amphiregulin and HB-EGF in ras- and myc-transformed embryonic fibroblasts from Tace ΔZn/ΔZn mice. However, as detailed in Table 1⇓, data for none of the other ligands or proteases provide the ideal composite of data to demonstrate loss of function in null cells, restoration by the candidate protease, and evidence of direct cleavage. In fact, in the case of ADAM9 as a candidate protease for the cleavage of HB-EGF, constitutive and stimulated ectodomain shedding of HB-EGF is comparable in embryonic fibroblasts isolated from ADAM9–/– and from wild-type mice, arguing against an essential role of ADAM9 in HB-EGF shedding in these cells (26).
The complexities associated with both ligand and protease processing also contribute to the difficulties in unambiguously assigning a role for a particular protease to processing of an identified ligand. As depicted in Figure 1⇓, ErbB ligands move through the secretory pathway where different modifications and cleavage events can take place. In Drosophila, the activity and release of the soluble ErbB-like ligand, Spitz, is regulated by two other proteins, Star and Rhomboid (13,27,28). Star chaperones the Spitz precursor in the endoplasmic reticulum and directs its transport to the Golgi complex (29). Once in the Golgi apparatus, Rhomboid, a novel intramembrane serine protease, cleaves the Spitz pro-protein to initiate secretion (30). By analogy to the regulation of Spitz processing and release, any perturbation that interferes with processing and trafficking of ErbB ligands could appear to inhibit ligand cleavage and release. The overexpression of protease-inactive ADAM mutants may induce such perturbation. As pointed out by Sunnarborg et al., heterozygous Tace +/ΔZn mice display a wild type phenotype indicating that protease-inactive mutants do not function as dominant-negative proteins. In light of the complex and poorly understood biosynthesis and trafficking of ADAMs in the secretory pathway, the studies using overexpression of ADAM mutants should be viewed with caution, and other substrates and ADAM mutants should be used as controls. Thus, understanding the regulation of biosynthesis and trafficking of both ErbB ligands and their candidate proteases will be critical to defining the rate-limiting steps in proteolytic release of ErbB ligands.
Most ADAMs implicated in ErbB ligand processing are ubiquitously expressed, raising the possibility that distinct ADAM specificity for ligand processing may reflect variations in signaling and regulation of ADAM proteolytic activity in different cell types. Although multiple activators of ErbB ligand shedding have been identified (23–,25), the proteases responsible for the shedding per se have not been completely characterized. For further progress to be made in understanding the mechanisms regulating these “sheddases,” the experimental criteria required to identify candidate metalloproteinase(s) must be clearly defined. Sunnarborg et al. have begun to address the role and specificity of TACE in ErbB ligand processing, although many questions remain.
Data demonstrating a critical role for the proteolytic cleavage of ErbB ligands in ErbB signaling have created a fundamental paradigm shift in our understanding of the biology of ErbB ligand precursors. Numerous in vitro and in vivo studies support the concept that sequential processing of membrane-anchored ErbB ligands provides a mechanism for exquisite control over the temporal and spatial presentation of ligands to receptors (31–,39). Thus, sequential processing can directly regulate the ability of a particular ligand to switch function from a potential juxtacrine mode to an autocrine or paracrine mode (8–,11) (Figure 1⇓). The studies by Sunnarborg et al. and others have established that the release of soluble ErbB ligands is a key upstream regulatory event in ErbB receptor signaling, and have thus identified protein processing as a target for therapeutic intervention (15–17,40–,43). For example, the use of either neutralizing antibodies to the ErbB1 receptor, or metalloproteinase inhibitors to effectively inhibit ErbB signaling, can inhibit the in vitro autocrine-dependent proliferation and migration of mammary epithelial cells (15). Additionally, in these mammary epithelial cells, autocrine presentation of a protease-cleavable ligand results in enhanced, persistent cell migration in a particular direction, whereas stimulation by constitutively secreted ligand results in a scattering response in these cells (41). Is there any physiological role for juxtacrine signaling by ErbB ligand precursors in vivo? Or is the primary role of the membrane-anchoring domains of these ligands the spatial regulation of release to appropriate cellular compartments? Answers to these questions will shape our paradigms of ErbB ligand signaling in the future.
Members of the Erbb Ligand Family Shed by Metalloproteinases
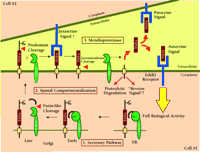
TACE-dependent proteolysis of the proTGFα extracellular domain as a regulatory event in ErbB1 receptor signaling. Genetic and biochemical data indicate that the release of soluble TGFα acting either as an autocrine or a paracrine signal is essential to achieve full biological activity in vivo. Data from Sunnarborg et al. suggest that TACE may also be involved in the N-terminal prodomain processing of proTGFα (16). Many questions still remain about the specificity and regulation of TACE as general ErbB ligand “sheddase.” Some of these questions can be divided into three general areas. 1. Secretory Pathway. How is the biosynthesis and activation of TACE regulated and can active TACE cleave substrate during transit through the secretory pathway? 2. Spatial Compartmentalization. What is the trafficking of TACE, what membrane compartments is it localized to and how is access of TACE to substrate regulated? 3. Metalloproteinases. What is the specificity of TACE for other ErbB ligands? Is the catalytic activity of TACE regulated? What is the role of other metalloproteinase(s) in ErbB ligand shedding?
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Kyle Garton, PhD, is currently finishing his MD training in the MSTP program at the University of Washington Medical School. The picture above was taken April 17, 2002, the day he completed his final examination for his Doctoral Degree.Elaine W. Raines, MS, is a Research Professor in the Department of Pathology at the University of Washington Medical School.

Peter J. Dempsey, PhD, is a Principal Scientist at Pacific Northwest Research Institute. Address comments to PJD. E-mail: PDempsey{at}pnri.org

