Small Interfering RNAs: A Revolutionary Tool for the Analysis of Gene Function and Gene Therapy
- 1Department of Cellular Biochemistry, Max-Planck-Institute for Biophysical Chemistry, D-37077 Goettingen, Germany
- 2Children’s University Hospital, Pediatric Hematology and Oncology, 35392 Giessen, Germany
- T.T. or A.B. E-mail ttuschl{at}mpibpc.gwdg.de; e-mail arndt.borkhardt{at}paediat.med.unigiessen.de
Abstract
RNA interference (RNAi) represents an evolutionarily conserved cellular defense for controlling the expression of foreign genes in most eukaryotes including humans. RNAi is triggered by double-stranded RNA (dsRNA) and causes sequence-specific mRNA degradation of single-stranded target RNAs homologous in response to dsRNA. The mediators of mRNA degradation are small interfering RNA duplexes (siRNAs), which are produced from long dsRNA by enzymatic cleavage in the cell. siRNAs are approximately twenty-one nucleotides in length, and have a base-paired structure characterized by two-nucleotide 3′-overhangs. Chemically synthesized siRNAs have become powerful reagents for genome-wide analysis of mammalian gene function in cultured somatic cells. Beyond their value for validation of gene function, siRNAs also hold great potential as gene-specific therapeutic agents.
The ability of double-stranded RNA (dsRNA) to silence the expression of genes has been the focus of many studies in C. elegans
and D. melanogaster. More recent research has looked for evidence of RNAi-mediated gene suppression in other model organisms.
Now there is excitement that RNAi-based methodologies will allow for the rapid assessment and validation of proteins as potential
drug targets. Additionally, we might now be standing on the edge of fundamentally new approaches to gene therapy as conducted
through RNAi-mediated suppression of mutated genes.
INTRODUCTION
When viruses infect eukaryotic cells, or when transposons and transgenes are randomly integrated into host genomes, dsRNA is frequently produced from the foreign genes. Most eukaryotes, including humans, possess an innate cellular immune surveillance system that specifically responds to the presence of dsRNA and activates processes that act post-transcriptionally to silence the expression of the interloping genes (1–4). This mechanism is now commonly referred to as RNA interference or RNAi (5). During RNAi, long transcripts of dsRNA are rapidly processed into small interfering RNAs (siRNAs), which represent RNA duplexes of specific length and structure that finally guide sequence-specific degradation of mRNAs homologous in sequence to the siRNAs (6, 7). The sequencing of the human genome has created an urgent need to ascertain efficiently the function of novel genes and to validate targets for drug discovery. Indeed, the rapid translation of the genomic DNA sequence information into therapeutic strategies for many common maladies—particularly infectious, cardiovascular, neoplastic, and neurological diseases—would be highly desirable. siRNAs may be the best tools for target validation in biomedical research today, because of their exquisite specificity, efficiency and endurance of gene-specific silencing. siRNAs are probably also suitable for the design of novel gene-specific therapeutics by directly targeting the mRNAs of disease-related genes.
The transfection of siRNAs into animal cells results in the potent, long-lasting post-transcriptional silencing of specific genes (8, 9). siRNA-mediated gene silencing is particularly useful in somatic mammalian cells, because these cells mount an additional, sequence-nonspecific innate immune response (i.e., responding with interferon-mediated defenses) when exposed to dsRNA greater than thirty base pairs, therefore prohibiting the application of longer dsRNAs (10). siRNAs are extraordinarily effective at lowering the amounts of targeted RNA—and by extension proteins—frequently to undetectable levels. The silencing effect is long lasting, typically several days, and extraordinarily specific, because one nucleotide mismatch between the target RNA and the central region of the siRNA is frequently sufficient to prevent silencing (6, 7, 11, 12). siRNAs can be rapidly synthesized and are now broadly available for the analyses of gene function in cultured mammalian cells. Similar to antisense oligonucleotide technology (13), the use of siRNAs also holds great promise for the application of gene-specific therapies in treating acute diseases such as viral infection, cancer, and, perhaps, acute inflammation.
The Mechanism of dsRNA Interference
A schematic illustration shows the mechanism of RNAi (Figure 1⇓). The key enzyme required for processing of long dsRNAs to siRNA duplexes is the RNase III enzyme Dicer, which was characterized in extracts prepared from insect cells, C. elegans embryos, and mouse cells (14–16). Dicer contains an N-terminal RNA helicase domain, a Piwi, Argonaute, Zwille/Pinhead (PAZ) domain (17), two RNase III domains, and a C-terminal dsRNA-binding motif. The PAZ domain is also present in Argonaute proteins, whose genes represent a poorly characterized family present in dsRNA-responsive organisms. Argonaute1 (AGO1) and Argonaute2 (AGO2), two of the five Argonaute proteins of D. melanogaster, appear to be important for forming the mRNA-degrading sequence-specific endonuclease complex, also referred to as the RNA-induced silencing complex (RISC) (18, 19). Dicer and AGO2 appear to interact in D. melanogaster Schneider 2 (S2) cells, probably through their PAZ domains; however, RISC and Dicer activity are separable, and RISC is unable to process dsRNA to siRNAs, suggesting that Dicer is not a component of RISC (18, 20). Possibly, the interaction between Dicer and AGO2 facilitates the incorporation of siRNA into RISC (20). The endonucleolytic subunit of RISC remains to be identified.
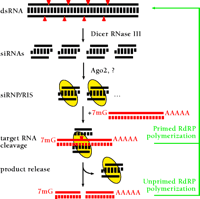
A model for RNA interference. dsRNA is processed to 21- to 23-nt siRNA duplexes by Dicer RNase III and possible other dsRNA-binding factors. The siRNA duplexes are incorporated into a siRNA-containing ribonucleoprotein complex (siRNP) (21) becoming the RNA-induced silencing complex (RISC) endonuclease, which targets homologous mRNAs for degradation. AGO2 and yet to be characterized proteins are thought to be required for RISC formation. The RISC mediates sequence-specific target RNA degradation. In plants and nematodes, targeted RNAs might also function as templates for double-strand RNA synthesis giving rise to transitive RNAi (24, 80–83), either through siRNA-primed dsRNA synthesis or through unprimed synthesis from aberrant RNA (which could represent the cleaved target RNA). In mammals or in fruitfly, however, RdRP genes have yet been identified, and the major mechanism of siRNA action is believed to be endonucleolytic target RNA cleavage guided by siRNA-protein complexes (RISC).
siRNA duplexes produced by the action of Dicer contain 5′-phosphates and free 3′-hydroxyl groups. The central base-paired region is flanked by two-to-three nucleotides of single-stranded 3′-overhangs (6). The 5′ -phosphate termini of siRNAs is essential for guiding mRNA degradation (21). Nevertheless, for their practical application in gene targeting experiments, siRNAs may be used without 5′-phosphate termini because a kinase activity in the cell rapidly phosphorylates the 5′ ends of synthetic siRNA duplexes (9, 21, 22). Under certain circumstances (e.g., injection experiments in D. melanogaster), 5′-phosphorylated siRNA duplexes may have slightly enhanced properties as compared to 5′-hydroxyl siRNAs (22). In gene targeting experiments using human HeLa cells, no differences in siRNA-mediated “knockdown” of gene expression were observed, as a function of 5′-phosphorylation (23). Furthermore, a cell line that is unable to utilize synthetic 5′-hydroxyl siRNAs for RNAi has not been encountered (for cell lines supporting RNAi see Table 1⇓).
siRNA-Mediated Silencing in Cell Lines
In C. elegans, introduction of approximately 300 bp dsRNA corresponding to a segment of the targeted gene may also give rise to the phenomenon of transitive RNAi (24). Transitive RNAi is characterized by the spreading of silencing outside of the region targeted by the initiator dsRNA. Presumably, targeted mRNA serves as template for RNA-dependent RNA polymerase (RdRP) and forms new dsRNA that is processed by Dicer. Thus, secondary siRNAs are generated that may cleave the mRNA outside of the region targeted by the ancestral dsRNA. Although this appears to have important implications for RNAi-based analysis of gene function because silencing may spread between genes that share highly homologous sequences, phenotypic analysis of a large set of silenced genes in C. elegans suggests that transitive RNAi between naturally occurring homologous gene sequences is probably of no major concern (25, 26). It was also proposed that siRNAs might prime novel dsRNA synthesis; however, it should be pointed out that siRNAs, in comparison to longer dsRNAs, are extremely poor initiators of gene silencing in C. elegans(27, 28). Biochemical evidence for RdRP activity in D. melanogaster was recently reported (29), although genes encoding classical RdRP activity appear to be lacking from the D. melanogaster genome. Despite the beauty of the suggested model in D. melanogaster, which hypothesizes that siRNAs function as primers for target-RNA-dependent dsRNA synthesis, thus leading to amplification of the silencing signal (29), biochemical evidence for the spreading of gene silencing outside of regions targeted by dsRNAs has not been observed in other model systems (6, 12, 23, 30, 31). Rather, the predominant pathway of gene silencing appears to be siRNA-mediated target mRNA degradation by RISC formation, which may also act catalytically. Similar to the situation in D. melanogaster, genes encoding RdRPs have not been identified in mammals. Therefore it is important to remember that the mechanisms of silencing differ between different species.
Application of siRNAs in Somatic Mammalian Cells
siRNAs have brought reverse genetics to mammalian cultured cells, and have made large-scale functional genomic analysis a realistic possibility (32). Standard cell lines provide starting points for mammalian functional screens because siRNAs can be effectively delivered by electroporation or cationic liposome-mediated transfection (11, 23). For small scale-applications, the microinjection of siRNAs may represent an alternative method. Technical problems that result from low transfection efficiencies may be partially overcome by using cell sorting protocols, such as after the transfection of siRNAs together with sorting markers such as GFP-expression plasmids. Alternatively, siRNAs that target cell surface marker proteins may be co-transfected, and the reduced expression of the co-targeted cell surface marker could then be used to identify specific cell populations by cell sorting.
An obvious prerequisite for the application of siRNAs for validation and therapeutic applications is the need for functional RNAi machinery within the targeted cells or tissue to bind to siRNAs and mediate mRNA degradation. In order to assay for the activity of this ribonucleoprotein complex in cells, a reporter assay was developed (9, 23). Plasmids coding for firefly and sea-pansy luciferase are transfected together with control or target-specific (i.e., luciferase) siRNAs into cells, and the relative luminescence of target versus control luciferase activity is measured.
siRNAs have been used to identify cytoskeletal proteins that are essential for cell growth (32). Even the targeting of non-essential genes resulted in cellular phenotypes that were identical to phenotypes previously observed in cells derived from transgenic knockout mice (32), illustrating the value of siRNA methodology for the analysis of mammalian gene function.
In certain situations, several-hundred-base-pair long dsRNA represents an alternative to siRNAs. Long dsRNA effectively silences genes expressed in insect cells (18, 33–35) and in embryonic mammalian cells that have not yet established the interferon system (15, 36–39). However, undifferentiated cells, such as embryonic stem (ES) cells or P19 teratocarcinoma cells, are difficult to work with because these progenitor cells are often poorly transfectable, making cell sorting prior to phenotypic analysis necessary (15).
Until recently, the application of siRNAs in somatic cells was restricted to the delivery of chemically or enzymatically synthesized siRNAs (9, 40–42) (Figure 2A⇓), but methods for intracellular expression of small RNA molecules have now been developed. Endogenous delivery is possible by inserting DNA templates for siRNAs into RNA polymerase III (pol III) transcription units, which are based on the sequences of the natural transcription units of the small nuclear RNA U6 or the human RNase P RNA H1. Two approaches are available for expressing siRNAs: 1) The sense and antisense strands constituting the siRNA duplex are transcribed from individual promoters (42–44), or 2) siRNAs are expressed as fold-back stem-loop structures that give rise to siRNAs after intracellular processing by Dicer (11, 41, 42, 45, 46) (Figure 2B⇓). The transfection of cells with plasmids that encode siRNAs, therefore, represents an alternative to direct siRNA transfection. Stable expression of siRNAs may facilitate certain applications such as the functional characterization of non-essential gene products in synthetic lethality screens or the construction of combinatorial libraries useful for loss-of-function screening in microarray assays (47). The insertion of siRNA expression cassettes into (retro)viral vectors will also enable the targeting of primary cells refractory to transfection or electroporation of plasmid DNA.
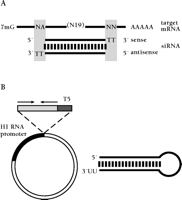
Methods for the delivery of siRNAs to somatic mammalian cells. (A) Synthetic 21-nt siRNA duplex prepared by chemical synthesis (23) aligned to a target mRNA. Target regions are selected such that siRNA sequences may contain uridine residues in the 2-nt overhangs. Uridine residues in the 2-nt 3′-overhang can be replaced by 2′-deoxythymidine without loss of activity, which significantly reduces costs of RNA synthesis and may also enhance nuclease resistance of siRNA duplexes when applied to mammalian cell (7). (B) Plasmid-based expression of short hairpin loops which give rise to siRNAs in vivo (11). The polymerase III promoter of H1 RNA (human RNase P RNA) drives the transcription of a nineteen-base-pair/nine-nucleotide-loop RNA hairpin. The transcription is terminated by the encounter of a polythymidine tract (T5) after the incorporation of two to three uridine residues encoded by the T5 element. Northern blot analysis showed that the hairpin RNAs were processed to siRNAs.
The function of several genes in cultured somatic mammalian cells have been analysed using siRNAs. The human vacuolar sorting protein Tsg101 was thus identified as essential for HIV-1 but not MLV budding (48). The reintroduction of a Tsg101-expressing plasmid that encoded Tsg101 mRNA with silent mutations at the siRNA-targeting site restored the HIV-1 budding defect. siRNA-mediated depletion of endogenous targets and the re-introduction of siRNA-resistant rescue constructs (30) will become important for the analysis of protein function much like the complementation analyses used in traditional yeast genetic research. In other instances, siRNAs were applied for studying the role of proteins involved in DNA damage response and cell cycle control (11, 49–53), general cell metabolism (54–56), signaling (57–59), cytoskeleton and its rearrangement during mitosis (32, 44, 60), membrane trafficking (61, 62), transcription (63), and DNA methylation (64). These various examples reported by independent investigators illustrate the robustness of the siRNA gene silencing technology.
However, variations do exist in the efficiency of siRNAs to target the same genes (12, 23, 32). Our experience in targeting many different genes suggests that, on average, between seventy to ninety percent of randomly chosen siRNAs are able to reduce target gene expression by more than eighty percent. Some genes, encoding extremely abundant and extremely stable proteins (e.g., vimentin), may be more difficult to silence, and the probability for finding efficacious siRNAs may be lower (23, 32). Nonetheless, difficulty in depleting the most abundant proteins does not appear to compromise the value of this new transient gene silencing technology.
siRNAs as Novel Therapeutic Platform Technology
siRNAs are highly sequence-specific reagents and discriminate between single mismatched target RNA sequences (7, 11), and may represent a new avenue for gene therapy. The expression of mRNAs coding for mutated proteins, which give rise to dominant genetic disorders and neoplastic growth, might be decreased or blocked completely by specific siRNAs.
With respect to targeting viral gene products expressed in virus-infected cells, it is possible that infectious mammalian viruses could express inhibitors of RNAi similar to those identified in plant and insect viruses (65–71). Because viral inhibitors of the mammalian RNAi machinery have not yet been described, it seems feasible that the application of siRNAs could extend our understanding of viral protein function and viral life cycle. Bitko and Barik successfully used siRNAs to silence genes expressed from respiratory syncytial virus (RSV) (72), a negative strand RNA virus that causes sometimes severe respiratory disease, especially in neonates and infants. They also demonstrated that siRNAs do not induce interferon-mediated responses, by showing the absence of phosphorylation of the translation factor eIF-2α. Although mRNAs transcribed from the viral genome were effectively silenced and viral replication was inhibited, it was not possible to cleave the viral genomic or antigenomic RNA because of its chromatin-like condensed structure. Lee et al. recently reported effective siRNA-mediated degradation of HIV-1 rev transcripts in a cell assay by co-transfection of proviral DNA and siRNA expression vectors, thus raising the possibility that siRNAs may become useful to treat HIV infection (43).
In leukemias and lymphomas—the most frequent cancers in childhood—oncogene activation frequently occurs through reciprocal chromosomal translocations. These translocations lead to juxtaposition of gene segments normally found on different chromosomes, and the creation of a composite gene. The prototype of such a translocation is the generation of the Philadelphia chromosome by the translocation of the long arms of chromosomes 9 and 22 [t(9;22)] in patients with chronic myelogenous leukemia and acute lymphoblastic leukemia (73). The translocation fuses the BCR gene from chromosome 22 and ABL gene from chromosome 9, creating an oncogenic BCR-ABL hybrid gene (Figure 3⇓) (74). The BCR-ABL fusion protein has dramatically increased the tyrosine kinase activity, as compared to that of the normal ABL protein, leading to aberrant phosphorylation of several downstream molecules. The kinase activities of both BCR-ABL and ABL can be inhibited by a specific tyrosine kinase inhibitor, STI 571 (Imatinib®), which is now used in the effective treatment of BCR-ABL-positive leukemia (75, 76). RNAi was also used to target the BCR-ABL mRNA, and this approach was compared to that of STI 571–mediated cell killing in a cell culture model. The siRNA treatment readily reduced the expression of BCR-ABL mRNA, followed by a reduction of BCR-ABL oncoprotein, leading to apotosis in leukemic cells (77). siRNA-based BCR-ABL silencing may become important considering that some patients develop drug resistance against STI 571 (e.g., by genomic amplification of BCR-ABL, increased expression of BCR-ABL mRNA or point mutation in the ABL gene). Alternative therapies, perhaps applied in combination with inhibitors such as STI 571, may help to overcome problems of such drug resistance.
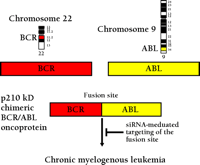
Scheme of the translocation t(9;22) in leukemia. The BCR-ABL fusion mRNA provides a leukemia-specific target that can be cleaved by siRNAs.
The combined effort of many laboratories worldwide has led to the molecular clarification of numerous chromosomal translocations through the successful cloning of the genes adjacent to the chromosomal breakpoint regions. Silencing of these tumor-specific, chimeric mRNAs by siRNAs might become an effective fusion gene-specific tumor therapy. The extraordinary sequence specificity of the RNAi mechanism may also allow for the targeting of individual polymorphic alleles expressed in loss-of-heterozygosity tumor cells, as well as targeting point-mutated transcripts of transforming oncogenes such as Ras. Finally, the decrease of overexpressed apoptosis inhibitors such as Bcl-2 and c-Myc might also be beneficial for cancer therapy. A list of possible targets for siRNA-mediated therapy in human malignancies is shown in Table 2⇓.
Selection of Possible Targets for Tumor Therapy by siRNAs
With respect to future medical applications, siRNAs were recently directed against a mutated mRNA associated with the spinobulbular muscular atrophy (SBMA) in tissue culture (78). SBMA, together with Huntington Disease, belongs to a growing group of neurodegenerative disorders caused by the expansion of trinucleotide repeats (79). Targeting the CAG-expanded mRNA transcript with dsRNA may be an attractive alternative to commonly used therapeutic strategies that, beyond symptomatic treatment, mainly focus on the inhibition of the toxic effects of the polyglutamine protein. Caplen et al. (78) successfully decreased the expression of mutated transcripts of the androgen receptor in human kidney 293T cells that were transfected with a plasmid encoding the expanded-CAG androgen receptor mutant. Most importantly, the authors achieved a rescue of the polyglutamine-induced cytotoxicity in cells treated with dsRNA molecules. Even though the study observed RNAi in transfected cells in vitro rather than in a more physiologically relevant context, the approach provides proof-of-principle, and underlines the remarkably broad potency and sequence-specificity of RNAi-mediated gene therapy. Whether the RNAi pathway is functionally active in various neuronal cells irrespective of their state of differentiation remains to be shown.
The delivery of siRNAs to the proper sites of therapy remains problematic. This is especially true for their delivery to primary cells, because such cells often do not tolerate treatment with liposome transfection reagents. Chemical modification of siRNAs, such as changing the lipophilicity of the molecule may be considered—for example, phosphorothioate modifications present in antisense oligodeoxynucleotides, or the attachment of lipophilic residues at the 3′-termini of the siRNA duplex. Delivery of siRNAs into organisms might be achieved with methods previously developed for the application of antisense oligonucleotides or nuclease-resistant ribozymes. Such methods consist of the injection of naked or liposome-encapsulated molecules. Studies that inform us about the possibility of exploiting RNAi in various cell types, tissues, and organs are urgently needed. Without doubt, these experiments will be performed in the near future in academic as well as industrial settings.
CONCLUSIONS
The pace with which siRNAs revolutionize the analysis of mammalian gene function is astounding, considering that siRNA-mediated gene silencing was only introduced last year (9, 78). siRNAs are poised to facilitate the genome-wide systematic analysis of gene function in cultured cells, and may soon become a valuable tool for target validation beyond in vitro tissue culture. siRNAs may yet provide a solution for gene-specific drug development, especially before highly specific small-molecule inhibitors become available.

Thomas Tuschl, PhD, is an EMBO Young Investigator and leads the Combinatorial Biochemistry Research Group at the Department of Cellular Biochemistry, Max-Planck-Institute for Biophysical Chemistry, Goettingen, Germany.

Arndt Borkhardt, MD, is a Principal Investigator in the Department of Pediatric Hematology and Oncology, at Children’s University Hospital, Giessen, Germany. Address correspondence to either T.T. or A.B. E-mail ttuschl{at}mpibpc.gwdg.de; e-mail arndt.borkhardt{at}paediat.med.uni-giessen.de
Acknowledgments
We acknowledge Sayda Elbashir, Jens Harborth, Klaus Weber and Ulrike Krämer for critical comments on the manuscript. Our own studies were made possible by grants from the Deutsche Forschungsgemeinschaft.
- © American Society for Pharmacology and Experimental Theraputics 2002

