Isoforms of Mammalian Adenylyl Cyclase: Multiplicities of Signaling
- 1The Department of Pharmacology University of Michigan Medical School Ann Arbor, Mi 48109-0632
- 2The Department of Pharmacology and the Alliance for Cellular Signaling University of Texas Southwestern Medical Center Dallas TX 75390-9041
- RKS or RT. E-mail sunahara{at}umich.edu; fax 734-763-4450. E-mail ron.taussig{at}utsouthwestern.edu.
Abstract
Mechanistic descriptions of cAMP production have evolved significantly since the 1960s when Sutherland and Rall hypothesized the existence of a single polypeptide that would both recognize hormone and synthesize cAMP. We now appreciate that the hormone-activated synthesis of cAMP involves multiple polypeptides, including a membrane-bound receptor; a heterotrimeric, guanine nucleotide–binding protein (G protein); and a membrane-bound adenylyl cyclase (AC). Biochemical and structural biological studies have provided a firm understanding for the regulation of AC by G proteins and elucidated the catalytic mechanism. In addition, a number of small molecules have been developed that modulate AC activity, introducing AC as a potential therapeutic target. Many paradigms of multi-modal regulation of AC have been investigated from a physiological perspective. This review addresses the complexity of the direct modulators of AC and summarizes the current biological models of their function.

With nine different isoforms of membrane-associated adenylyl cyclases (ACs) and one isoform of soluble AC, there is much to
learn and even more to understand regarding the expression of tissue-specific AC isoforms. However, on the protein level,
there are many proteins and small molecules that affect the catalytic activity of ACs. Knowing how to tailor AC activity,
or how to exploit the activity of one isoform over another in a given tissue, may give rise to therapeutic agents that can
inhibit AC-dependent disease states or, at least, lessen their severity.
INTRODUCTION
The activation of adenylyl cyclase (AC), resulting in the intracellular production of adenosine-3′,5′-monophosphate (i.e., cyclic AMP [cAMP]), is initiated by the binding of hormones to cell surface receptors (1). Epinephrine, dopamine, prostaglandin PGE2, adenosine, and glucagon are a few examples of the many hormones that activate AC through membrane-bound receptors. Glucagon, for example, a hormone that regulates glycogen metabolism in liver and skeletal muscle, recognizes membrane receptors in these tissues, markedly stimulates AC to produce intracellular cAMP. Glucagon-bound receptors communicate with an intracellular, membrane-associated heterotrimeric G protein (2) composed of a guanosine diphosphate (GDP)–bound α-subunit and an obligate βγ heterodimer. Hormone-dependent activation of receptors leads to the exchange of GDP for guanosine triphosphate (GTP). Conformational changes due to GTP binding result in the dissociation of the heterotrimeric G protein into α and βγ subunits, which then interact with their respective effectors (Figure 1⇓).

G proteins mediate the effects of hormone signals on adenylyl cyclase.
A. Hormones that affect intracellular adenylyl cyclase activity bind to protein receptors that contain seven transmembrane domains (blue) and are thus anchored at the cell surface (brown double bar). The intracellular portion of these receptors interacts with GDP-bound heterotrimeric G proteins, resulting in the displacement of the bound GDP by GTP and concomitant dissociation of the GTP-bound Gα-subunit from the Gβγ-dimer.
B. Two families of Gα exist: GTP-bound Gαs stimulates adenylyl cyclase, whereas GTP-bound Gαi inhibits adenyly cyclase. Gα subunits possess an intrinsic GTPase activity, so that their dissociation from Gβγ and their effect on AC activity are transitory. (Yellow triangle represents GDP; red triangle represents GTP.)
There are multiple classes of α-subunits that regulate AC, either in a stimulatory (Gαs family), or inhibitory (Gαi family) manner (2). The two Gα families are normally coupled to distinct receptor subtypes. The βγ-subunits also regulate AC, but in an AC subtype–specific manner (3–5). Additionally, calcium ions are very strong modulators of some isoforms of AC (6–9); thus, G proteins that regulate calcium entry through voltage-dependent Ca2+ channels may also regulate AC activity (10–12).
ACs have been extensively characterized, and great advances have been made in our understanding of how they function. Their mechanism of action can now be incorporated into model systems to explain drug responses that occur in tissues or whole organisms. For example, learning and memory are associated with the activation of protein kinase activity and protein phosphorylation (13), two processes that are strongly regulated by AC activity. Genetically modified mouse strains that contain altered AC genes display considerable behavioral defects, particularly in learning and memory (14–16). The effects of Ca2+ and Ca2+–calmodulin (CaM) on AC activity are also strongly implicated in learning and memory (17–19). AC activity itself can be modulated through phosphorylation of the enzyme, and alterations in the expression of AC isoforms also accompany drug-induced receptor effects that have been related to symptoms of drug dependence (20–23).
Much of the research surrounding cAMP signaling has focused on AC activity that is regulated by G proteins. However, a bona fide soluble form of AC that is insensitive to G proteins and forskolin has been cloned (24). Soluble AC (sAC) activity was identified in testis during the early 1980s, but the activity remained an enigma until the corresponding cDNA was isolated. Elevated concentrations of cAMP in the testis are crucial for sperm development and capacitation. The discovery of sAC, along with the finding that it is activated by bicarbonate, has forced a reexamination of how cAMP signals are propagated into the cell beyond the cell membrane.
MULTIPLE AC ISOFORMS
Molecular cloning techniques have identified nine mammalian genes that encode membrane-bound ACs (3–5, 25), and one gene encoding a soluble isoform (24). These genes do not tend to cluster within the genome, but rather are distributed among different chromosomes, with the exception that the two genes that encode AC7 and AC9 are located on chromosome 16, albeit at opposites arms. The ten AC isoforms can be divided into five distinct families based on their amino acid sequence similarity and functional attributes. The Ca2+–CaM-sensitive forms are types AC1, AC3, and AC8 (Figure 2A⇓). The Gβγ-stimulatory forms are represented by AC2, AC4, and AC7 (Figure 2B⇓). AC5 and AC6 are distinguished by their sensitivity to inhibition by both Ca2+ and Gαi isoforms (Gαo, Gαi1, Gαi2, Gαi3, and Gαz) (Figure 2C⇓). AC9 is the most divergent of the membrane-bound family and is highly insensitive to the diterpene forskolin. The last isoform, sAC, is the most divergent of all the mammalian cyclases, and is similar to cyclases found in cyanobacteria (24).
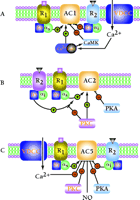
Multiple modes of regulation of adenylyl cyclase isoforms. (A) The pattern of regulation of AC1 as illustrated is representative also for AC3 and AC8. R1 represents a G protein–coupled receptor, such as the glucagon or β2-adrenergic receptor, that couples to the stimulatory G protein Gαs. R2 represents a G protein–coupled receptor, such as the muscarinic M2 or α1-adrenergic receptor, that couples to the inhibitory G protein Gαi. (B) The pattern of regulation of AC2 as illustrated is representative of the regulation of AC4 and AC7. Note that Gβγ regulation of AC2 is dependent on Gαs co-activation and does not activate AC by itself. PKC can use AC as a substrate, resulting in elevation of basal activity and inhibition of the Gβγ superactivation. (C) The pattern of regulation of AC5 is representative also of AC6. (PKA, protein kinase A; PKC, protein kinase C; CaM, calmodulin; CaMK, calmodulin-dependent kinase; NO, nitric oxide; VDCC, voltage-dependent Ca2+ channel.)
The distribution of these AC isoforms, according to mRNA detection, is summarized in Table 1⇓. In general, all membrane-bound AC isoforms are found in, but not limited to, excitable tissues such as neurons and muscle (3, 26). Within the brain, AC isoforms localize to different, discrete brain regions (27–29). Although most isoforms are widely expressed, AC1 and AC3 are expressed only in brain (29). Soluble cyclase is expressed predominantly in the testis, although splice variants have been identified displaying a broader distribution pattern (30). The broad distribution of AC isoforms suggests that any given cell contains multiple isoforms.
Regulatory Properties of Mammalian Adenylyl Cyclase Isoforms
REGULATION OF AC ACTIVITY
Stimulation by Gαs
Hormonal activation of AC occurs primarily through receptors coupled to the stimulatory G protein Gαs. Gαs is the most widely distributed activator of all mammalian membrane-bound AC isoforms. Multiple splice variants of Gαs have been identified: Gαs-short, Gαs-long and GαsXL. Although the former two isoforms have been extensively characterized both physiologically and biochemically, GαsXL is a relatively new member and is less well characterized (31). The long and short splice forms are biochemically indistinguishable in their capacity to directly activate AC (32); however, the behavior of the hormone receptor–stimulated AC varies considerably (33). GαsXL can activate AC directly, but no hormone receptor–mediated effects through GαsXL have been demonstrated (34).
Gαs in the GTP-bound form displays a tenfold greater affinity for activating AC compared to the GDP-bound form (35). Crystallographic evidence suggests that the main contact between Gαs and AC occurs through a short α-helix that is highly mobile throughout the GTPase cycle of all G proteins (36, 37). The decreased affinity of the GDP-bound form for AC suggests that the GTPase activity of Gαs serves as a timing mechanism to delimit cyclase activation. Following GTP hydrolysis, Gαs dissociates from cyclase, reassociates with Gβγ, and thereby terminates both Gαs and Gβγ signaling. The deactivation of Gαs can be accelerated by a specific Regulator of G protein Signaling (RGS) molecule, PX1-RGS, that serves as a GTPase-accelerating protein (GAP) for Gαs (38). AC itself can weakly accelerate the GTPase activity of Gαs (39), similar to the accelerating effect of PLCβ isoforms on their activator Gαq (40).
Inhibition by Gαi
Members of the Gαi family inhibit AC but can manifest selectivity for given AC isoforms. Gαi1, Gαi2, Gαi3, Gαo, and Gαz can inhibit AC5 and AC6 (Figure 2C⇑) (41–43). Interestingly, their mode of inhibition is not through direct competition with Gαs, because forskolin-stimulated activity is also inhibited. In addition, mutagenesis experiments and structural modeling suggest that Gαi exerts its effects at a site, symmetrical to the Gαs binding site, located on the side opposite the AC molecule (44). The highly expressed brain-specific Gαo can inhibit AC1 (and possibly AC8), although it is not as potent as the other Gαi subunits on AC5 and AC6. The G αi subunits are posttranslationally modified by long-chain acyl (myristoyl) and thioacyl (palmitoyl) moieties (45); myristoylation is required for Gαi-mediated inhibition of AC.
Regulation by Gβγ
The contributions of the Gβγ heterodimer to the modulation of ACs have been, at least until recently, largely unappreciated (46). G α-subunits had long been presumed to predominate in the regulation of AC; however, Gβγ subunits are strong modulators of AC activity that can either be stimulatory, as in the case of AC2, AC4, and AC7, or inhibitory, as for AC1 and AC8 (Figure 2⇑) (46, 47). In fact, Gβγ subunits are among the most potent of all negative regulators of AC1 and AC8, and can markedly inhibit the effects of forskolin, Gαs, and Ca2+–CaM on AC activities. These findings are particularly relevant for brain physiology because the Gαi family and their accompanying βγ subunits are, along with AC1 and AC8, highly expressed in the brain (48).
In contrast, Gβγ subunits act to stimulate the cyclase activity of AC2, AC4 and AC7, albeit only when Gαs is co-activated (Figure 2B⇑). Gβγ and Gαs could thus establish a synergistic relationship, whereby the presence of Gβγ might dramatically enhance the ability of Gαs to activate AC. Indeed, the activation of those hormone receptors coupled to Gαi subunits could liberate Gβγ dimers that could synergistically potentiate AC activity that had been stimulated by distinct, Gαs-activated, hormone receptors. It is important to note that the AC isoforms that undergo stimulation by Gβγ (i.e., AC2, AC4 and AC7) are not directly modulated by the α subunits of the Gi family (3, 26). Less well understood is the relationship between Gβγ and the other cyclase isoforms such as AC5 and AC6. Transfection experiments suggest that Gβγ can inhibit AC5 and AC6 activity, perhaps in an indirect manner (49).
The putative binding site for Gβγ on the Gβγ-stimulated family of ACs (i.e., AC2, AC4, and AC7) has been mapped on the basis of peptide inhibition studies (50, 51). Peptides corresponding to amino acid residues 956 to 982 of AC2, that is, derived from the middle of the second of the two catalytic domains (i.e., C2), potently inhibit the ability of Gβγ to stimulate the enzyme activity of intact AC2. Despite the high degree of sequence conservation among AC catalytic domains, this sequence (i.e., corresponding to residues 956 to 982 of AC2) is not found in AC isoforms that are not modulated by Gβγ. Indeed, this sequence also contains the short putative Gβγ-binding motif QXXER, the consensus for which is based on GRK2, the β-adrenergic receptor kinase that requires Gβγ for activation, as well as Gβγ-activated inwardly rectifying K+ channels, the Gβγ-activated PLCβ isoforms, and the Gβγ-inhibited AC1. Disruption of the consensus QXXER motif in any of these instances abrogates all Gβγ effects. The C2 domain of AC2, possessing the QXXER motif, is located near the plasma membrane face, but the precise structure of this motif is unknown because the region is disordered in the crystal structure. An additional region within the regulatory region of the C1 domain, juxtaposed to the transmembrane domain, may also be important for Gβγ regulation of AC2, AC4, and AC7 (52).
A peptide generated from the catalytic region of the AC1 isoform analogous to that containing the QXXER motif in the AC2 sequence also displays dramatic effects on Gβγ regulation of AC activity (52). The peptide could reverse both Gβγ-dependent inhibition of AC1 activity and Gβγ-dependent superactivation of Gαs-stimulated AC2, suggesting that the region of the AC1 isoform also serves for binding of Gβγ. For example, it has been reported that this region contributes to Gβγ-mediated inhibition of AC1 activity (53).
The recent findings outlined above underscore the importance of the Gβγ heterodimer in modulating AC activity and suggest that it may be naïve to regard the importance of Gβγ as secondary to that of Gα. In addition to its role in regulating AC, Gβγ subunits have been implicated as the primary component of G proteins that directly regulate ion channels (e.g., K+-channel activation and Ca2+- and Na+-channel inhibition) as well as other effectors systems: GPCR kinases (activation), phospholipase Cβ isoforms (activation), and the mitogen-activated protein (MAP) kinase pathway (activation) (10, 54). The MAP kinase-dependent mating response in yeast is solely dependent on Gβγ for signaling and only requires the Gα subunit for its inactivation.
Calmodulin and Ca2+as Regulators
Ca2+–CaM activates isoforms AC1, AC8, and possibly AC3 (Figure 2A⇑) (55–57). Some, but not all, agents that elevate local Ca2+ levels in intact cells may thus dramatically enhance the activity of these isoforms. Specifically, intracellular Ca2+ from IP3-sensitive stores are unable to affect these Ca2+-sensitive AC isoforms, whereas activation of Ca2+ entry through voltage-gated Ca2+ channels or through capacitative entry is effective at activating these AC isoforms (58, 59).
Although millimolar (i.e., non-physiological) concentrations of Ca2+ inhibit all AC isoforms (Figure 3C⇓), AC5 and AC6 are inhibited by concentrations of Ca2+ in the μM range, well within the dynamic range of intracellular levels (Figure 2C⇑) (60); Ca2+ from capacitative entry are thought to be the sole physiological source of Ca2+ to inhibit the AC5 and AC6 (6). The fact that these AC isoforms are restricted mostly to brain-specific and excitable cell types, and are compartmentalized with voltage-gated Ca2+ channels, is consistent with this notion (Table 1⇑).
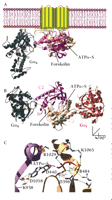
Structure of membrane-bound mammalian adenylyl cyclase bound to the activator Gαs. (A) Illustration of the crystal structure of the catalytic domain of adenylyl cyclase bound to Gαs (36) and superimposed onto the membrane-spanning region of mammalian adenylyl cyclase. Gαs•GTPγ–S in its activated form is demarcated in gray. The cyclase domains, C1 (tan) and C2 (mauve) interact and form the binding sites for forskolin and the substrate, ATP. (B) The same structure as in (A) but rotated around the x-axis of vision to give a perspective from the inner surface of the membrane. Gαi (in red) is overlaid onto the Gαs•C1•C2 structure at the pseudosymmetrically related binding site to the Gαs•GTPγ–S site. (C) The active site of adenylyl cyclase bound to the ATP analog ATPα–S(RP). Highlighted are residues that make contact with the nucleotide and that are conserved in all mammalian adenylyl cyclases. Asp396 (D396), Asp440 (D440), and Arg484 (R484) are in the C1 domain of AC5. Lys938 (K938), Asp1018 (D1018), Arg1029 (R1029), and Lys1065 (K1065) are in the C2 domain of AC2. Also indicated are two Mg2+ ions liganded by the phosphates of ATPα‴S(RP) and the two aspartate residues. The 3D structure was visualized with SwissPDBViewer™(178) and rendered with POV-RAY™ using coordinates from the Gαs•C1•C2•forskolin•ATPα–S(RP) (PDB id:1CJK) and Gαi (PDB id:1GIA) structures.
Regulation by Other Proteins
A number of other proteins have recently been identified that interact directly with ACs, but their biological significance has yet to be determined. Several of these proteins have been identified through yeast two-hybrid or copurification experiments using the cytosolic domains as baits. The protein associated with Myc (PAM) potently inhibits AC1 and AC5, but not AC2 (61), whereas the Escherichia coli protein SlyD, a cis-trans peptidylprolyl isomerase (PPIase), copurifies with bacterially expressed AC7 so as to inhibit its activity (62). A conceptually more relevant interaction was identified between another RGS protein, RGS2, and AC3 (63). RGS molecules are mostly noted for their ability to accelerate the GTPase activity of the heterotrimeric G proteins of the Gαi, Gαq, and Gα12 types. The RGS2 isoform can enhance the intrinsic GTPase rate of both the Gαi and Gαq (64, 65). A direct association between Gαi or Gαq and AC3 has not been demonstrated; however, this interaction would provide an additional avenue of crosstalk.
Regulation by Small Molecules
Forskolin
The diterpene forskolin (from Coleus forskohlii) potently activates all known isoforms of mammalian membrane-bound ACs with the exception of AC9 (66). The sensitivity difference may be accounted for by as few as two residues, Ala1112 and Tyr1082, corresponding to Leu912 and Ser942 of AC2 (67). In an unexpected divergence of evolution, however, the D. melanogaster ortholog of AC9 is sensitive to forskolin (68). The forskolin-dependent activation of AC2, AC4, AC5, AC6, and AC7 is synergistic with Gαs-mediated coactivation, whereas activation by forskolin and Gαs is additive for isoforms AC1, AC3, and AC8 (3).
The binding site for forskolin is located within the catalytic core of AC, at the interface between the intracellular catalytic (C1 and C2) domains (Figure 3⇑). Gαs binds similarly between the two domains, but at a location on the perimeter of the catalytic core. The relationship between the two binding sites and their proposed mechanism of action may explain the cooperativity of binding observed between forskolin and Gαs. Why other isoforms display additive effects with forskolin and Gαs is not obvious from the crystal structure. Stimulation of activation by forskolin and Ca2+–CaM is cooperative in the cases of AC1, AC8, and presumably AC3 (56, 69).
Since the elucidation of the crystal structure of AC bound to either forskolin or its water-soluble analog 7-deacetyl-7-(O-N-methylpiperazino)-γ-butyryl forskolin (5, 43), researchers have attempted to design isoform-selective forskolin analogs. Although this methodology is still in its infancy, several compounds have been synthesized that contain subtle modifications of forskolin and display a two- to threefold preference for certain cyclase isoforms (70).
Pyrophosphate
Adenylyl cyclase hydrolyses ATP to produce pyrophosphate and cAMP. In a steady-state AC assay, the rate-limiting step is normally the release of pyrophosphate (71). Elevated concentrations of pyrophosphate can thus be used to force AC into a product-bound conformation that prevents the binding of ATP. The antiviral agent, foscarnet, or phosphonoformic acid, mimics pyrophosphate and likewise inhibits AC activity (72).
P-Site Inhibitors
One collection of adenosine analogs, classified as P-site inhibitors, inhibits AC activity in a manner without competing with ATP binding (71, 73, 74). These compounds inhibit AC by binding to a conformation of the enzyme that closely resembles the product-bound state, or posttransition state (75). The capacity of P-site inhibitors to inhibit AC activity is thus dramatically affected by the catalytic activity of cyclase itself, which in turn is a function of the enzyme’s conformational state (73); most notably, P-site inhibitors are dramatically potentiated by the presence of pyrophosphate. The majority of P-site inhibitors lack one or more hydroxyl groups relative to the ribose ring structure (75). Additionally, most of these inhibitors are mono- or polyphosphates and are structural analogs of cAMP. Thus, 2′-deoxy-3′-AMP (IC50 ∼10 μM), and the more potent inhibitors 2′,5′-dideoxy-3′-ADP and 2′-5′-dideoxy-3′ATP (IC50 ∼ 40 nM), inhibit AC by stabilizing the quasi-product–bound state (76). P-site inhibitors are generally not specific for individual AC isoforms. The only exceptions are 9-(cyclopentyl)-adenine and 9-(tetrahydro-2-furyl)-adenine; they are ineffective on AC2, but equally inhibit AC1, AC3, AC5, AC6, AC7, and AC8 (70, 77).
Other Small-Molecule Modulators of AC Activity
Potent inhibitors of AC activity include the RP stereoisomer of α-thio-ATP (IC50 ∼ 1 μM), although the SP isomer is actually a weak inhibitor (78). α,β-Methyleneadenosine-5′-triphosphates (AMP-CPP), which contains a methylene group between the α- and β-phosphates, is also an effective inhibitor of AC activity (IC50∼300 μM) (79). The most potent inhibitor of AC activity currently available is β–L-2′,3′-dideoxy-5′-ATP (IC50 ∼ 24 nM) (80). As is the case with other inhibitors, such as 9-(2-phosphonylmethoxyethyl)-adenine and derivatives (80, 81), the ability of any these compounds to specifically modulate AC isoforms is not established.
Regulation by Posttranslational Modification
Several modes of posttranslational modification, including phosphorylation, glycosylation, and S-nitrosylation, can alter the activity of ACs. The phosphorylation of AC by protein kinases generally has an inhibitory effect, not on basal activity, but on enhanced stimulation by various activators. These effects are part of a negative feedback mechanism; for example, PKA-mediated phosphorylation is thought on negatively regulate AC5 and AC6 activity (Figure 2C⇑)(82, 83).
Much attention has focused on the role of phosphorylation by protein kinase C (PKC) in regulating AC activity since the initial report that AC purified from brain can be directly phosphorylated by this kinase (84). The activities of AC1, AC2, AC3, and AC5 can be stimulated following phorbol ester treatment, whereas those of AC4 and AC6 are inhibited, suggesting that PKC can regulate ACs in an isoform-specific manner (85–90). For AC2, AC5, and AC6, this regulation is due to direct phosphorylation by PKC (87, 91). Interestingly, although PKC has opposite effects on the Gαs-stimulated activities of AC2 (enhanced by PKC) and AC4 (inhibited by PKC), PKC causes both AC2 and AC4 to lose responsiveness to the (stimulatory) effect of Gβγ. In this way, PKC bears the role, with regard to AC2-like cyclases, of modulating the integration of Gαs and Gβγ inputs (85).
It is perhaps counterintuitive that Ca2+–CaM, which normally activates AC1, AC3, and AC8, can also inhibit AC1 and AC3 indirectly through phosphorylation by CaM kinase II and IV, respectively (92, 93). This mode of regulation most likely reflects a negative feedback loop that controls Ca2+-mediated stimuli.
Both hormone- and forskolin-stimulated AC5 and AC6 activity are inhibited by nitric oxide (NO) (94). In addition to its primary target—soluble guanylyl cyclase (GC) (95, 96)—NO affects the ryanodine receptor and the NMDA receptor (97–98). These effects are largely inhibitory and involve S-nitrosylation. More recently, N-linked glycosylation was demonstrated to be important for AC responsiveness to hormones and forskolin (99, 100). Although tunicamycin treatment, or substitution of glutamine for Asn805 and Asn890 of AC6, has very little effect on the targeting of AC6 to the plasma membrane and on G protein activation, G protein–mediated inhibition and responses to forskolin are impaired by as much as fifty percent (99). In contrast, a variant of AC8 requires N-linked glycosylation for plasma membrane targeting and thus for activation by membrane-bound G protein–coupled receptors (100, 101).
STRUCTURE: PRIMARY, SECONDARY, AND TERTIARY
AC is an integral membrane protein composed of twelve transmembrane segments. The protein can be visualized as two tandemly repeated domains, each containing six transmembrane segments and a large cytoplasmic (catalytic) loop (Figure 4⇓). The twelve-transmembrane domain topology is reminiscent of the ABC family of transporters such as the cystic fibrosis transmembrane rectifier and the P-glycoprotein, which is responsible for multidrug resistance and is encoded by the MDR1 gene (102, 103). The sequence similarity between the two cytosolic domains is striking: approximately forty percent, over a span of 250 residues, regardless of which membrane-bound isoform is considered (3). The catalytic cytosolic regions of mammalian ACs also share significant sequence similarity to the corresponding regions of GCs (96) and ACs from prokaryotes (Figure 4⇓).
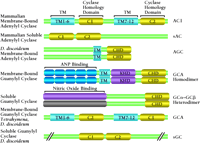
Sequence alignment of the adenylyl and guanylyl cyclases. The catalytic domains (yellow) display considerable similarity in amino acid sequence and have been coined the Cyclase Homology Domain (CHD). Illustrated in light blue are the membrane-spanning regions as predicted from amino acid sequence. (TM, transmembrane; ANP, atrial natriuretic factor; KHD, kinase homology domain.) Adapted from Wedel and Garbers (96).
The biochemical characterization of recombinant forms of the AC cytoplasmic domains has provided extremely useful insights into AC regulation and catalysis. The construction of fused C1–C2 domains (104) and individual soluble domains (35, 105–108) results in activities characteristic of the full-length membrane-bound AC forms in terms of modulation by G protein α-subunits, forskolin, substrate inhibitors, and P-site inhibitors (35, 105–108). The utilization of the recombinant, soluble domains of AC has facilitated the biophysical characterization of enzyme function.
Structural Basis for the Regulation of AC
The C1 and C2 domains that form the catalytic core of AC are related by two-fold pseudosymmetry (Figure 3⇑). Current models of the AC structure are based on the forskolin-bound form in the presence (C1–C2 heterodimer) or absence (inactive C2 homodimer) of Gαs. The forskolin binding site is located in a hydrophobic pocket at the interface between the two domains (36, 109). Like forskolin, Gαs also contacts both domains, with most of the binding surface (approximately seventy-five per cent) contributed by the C2 domain. The binding of Gαs induces a 7° rotation of the C1 domain around the C2 domain, presumably positioning the active site for catalysis. The C2 domain contacts Gαs primarily in the switch II region, one of three segments of G proteins that are highly mobile throughout the cycle of GTP hydrolysis (37). The pseudosymmetrical structure of the catalytic core makes apparent the likely binding site for other G proteins, such as Gαil, that regulate catalysis (Figure 3B⇑). Gαi selectively inhibits AC5 and AC6, for example, presumably by binding to the C1 domain (and perhaps through an interaction with the C2 domain) and stabilizing the two helices of C1, thus allosterically modifying the proximally located active site (44, 108).
Substrate Binding and the Mechanism of Hydrolysis
The active site, revealed by x-ray diffraction of C1•C2•Gαs•forskolin co-crystals, is located at the interface between the C1 and C2 domains in at a site pseudosymmetrically related to the forskolin binding site (36, 109, 110). Residues that contact the substrate (or substrate analog) are conserved in all AC isoforms (Figure 3C⇑). Interestingly, the positions of Lys938 and Asp1018 in AC2, residues that contribute most of the binding energy to the adenine ring, are occupied in GCs by glutamate and cysteine residues, respectively. Indeed, the substrate specificity of AC can be changed from ATP to GTP by making the appropriate amino acid substitutions (111). Similarly, the conversion of a GC to an AC has also been demonstrated (111, 112).
The residues that coordinate the binding of the ribose and triphosphate portion of the nucleotide are conserved in all isoforms AC and GC (Figure 3C⇑). Non-conservative substitution of any of these residues severely impairs cyclase activity. Arg484, Arg1029, and Lys1065 in AC2 share coordination of the α-, β-, and γ-phosphates of the nucleotide. Two highly conserved aspartate residues (Asp396 and Asp440 in AC5) also help to coordinate the phosphates by coupling to two Mg2+ cations that stabilize the α-phosphate during catalysis. The overall structure is strikingly similar to that found in other phosphoryl transferases, such as T7 DNA polymerase and HIV reverse transcriptase (110, 113–115). A model of the catalytic mechanism for these enzymes involves the contribution of one the Mg2+ ions acting as a general base that deprotonates the 3′-OH of the ribose ring (116). The newly formed oxyanion is thus poised for nucleophilic attack of the α-phosphate with elimination of pyrophosphate.
PHYSIOLOGY AND FUNCTION OF MAMMALIAN ACs
The biochemical assessment of the ACs has revealed several regulatory pathways that control AC activity. In contrast, a number of factors have resulted in a relative paucity of physiological data describing these complex regulatory pathways. The most notable obstacle is the multiplicity of the isotypes expressed within a given cell type, further complicated by the varying effects of modulators such as Ca2+ and Gβγ, as well as the particular intracellular milieu. The majority of data come form: 1) overexpression studies using cell transfection or transgenic animals; 2) gene disruption studies utilizing genetic knockouts; and 3) the identification of natural gene mutations.
Sensitization
The importance of AC sensitization has long been appreciated in model systems for drug abuse, withdrawal, and recovery (117). Cells chronically treated with opiates (which activate Gαi-coupled receptors) exhibit AC activity that is supersensitive to stimulation by either forskolin or Gαs following withdrawal of the opiate. Similar sensitization is observed with chronic activation of other hormone receptors that couple through Gαi—such as the A3 adenosine (118), D2 and D4 dopamine (23, 119), and M2 muscarinic receptor subtypes (23, 120)—and is dependent on the expression of particular ACs. In transfection studies, sensitization, in the form of superactivation, is observed for AC1, AC5, AC6, and AC8, but not for AC2, AC3, AC4, or AC7 (21, 119, 120), but the mechanisms underlying this form of sensitization remain obscure. Interestingly, chronic opioid treament leads to relative desensitization of the AC2, AC4 and AC7 isoforms, which appears to be regulated through Gαs, Gαi, Gβγ, and PKC (119, 120, 121–126). PKC-mediated phosphorylation of AC5 increases AC activity in vivo and in vitro (127). Chronic hormone stimulation leading to higher steady-state amounts of PKC-mediated phospho-AC5 may account for some of the apparent sensitization. Gβγ has been implicated in the cannabinoid (CB1) receptor-mediated superactivation of AC1, AC3, AC5, AC6, and AC8, but not the ACs that are normally activated by Gβγ (128).
Supersensitization might result as an effect of increased expression of specific AC isoforms, PKA, and the cAMP-responsive element binding protein (CREB) (117, 129). In sharp contrast, chronic exposure to ethanol reduces such expression and leads to AC desensitization (130), suggesting that ethanol dependence greatly relies on the cAMP signaling pathway (131).
Genetic Manipulation of AC Isoforms and the Mouse Model
Much information on the physiological role of specific AC isoforms has come from studies on genetically altered animals. The role of CaM-regulated ACs in learning and memory has been hypothesized ever since the discovery that the basis for the learning defects in the Drosophila mutant rutabaga is an inactivating mutation of a CaM-activated AC (132). Double knockout mice deficient in both of the CaM-stimulated ACs, AC1 and AC8, exhibit neither long-term memory nor late long-term potentiation (133). Each of the single knockouts is normal in these functions; however, they display other neurological defects. These results emphasize the involvement of cAMP signaling pathways in pattern formation of the brain and provide definitive evidence for roles of the CaM-regulated ACs in higher brain function.
Studies on AC3-deficient mice demonstrate a critical role for AC3 in olfaction. These mice fail several olfaction-based behavioral tests, and lack electro-olfactogram responses elicited by either cAMP or IP3, despite the presence of other AC isoforms in olfactory cilia (134). These knockout mice also implicate AC3 as an important integrator of growth-inhibitory signals that stimulate cAMP formation and that inhibit the growth of arterial smooth muscle cells (135).
Mice that overexpress ACs provide additional insight into the physiological roles of specific isoforms. For example, the overexpression of AC7 in the central nervous system enhances acute responsiveness and tolerance to morphine (136). In disagreement with cell transfection data, the transgenic AC7 mice are also supersensitive to Gαs responses following morphine treatment. The cause of this discrepancy is unknown. Studies on transgenic mice overexpressing either AC5 or AC6 demonstrate important differences between these two prominent isoforms in the heart (137–138). Among the major differences are the cardiac β-adrenergic–dependent regulation of heart rate and contractility responses, and the cardioprotective effects of AC6, but not AC5, observed in mouse models of heart failure (induced by overexpression of Gαq).
Mutations of the AC System in Human Disease
A number of studies have associated impairments of AC systems with certain human diseases. Mutations causing constitutively active receptors—resulting in elevated intracellular cAMP concentrations—have been found in patients with: familial male precocious puberty/testitoxicosis (emanating from a constitutively activate mutant luteinizing hormone receptor) (139); overactive thyroid adenomas and non-autoimmune autosomal dominant hyperthyroidism (arising from excessive activation of thyroid-stimulating hormone receptor) (140); and Jansen-type metaphyseal chondrodysplasia (resulting from a constitutively active mutant parathyroid hormone receptor) (141). Similarly, diseases associated with mutations yielding constitutively active G proteins (Gαs) are found in patients with endocrine tumors, McCune-Albright syndrome, and testitoxicosis (142–144). Because elevated cAMP concentrations in isolated endocrine tumors can arise independently of oncogenic mutations of the G protein α-subunit, moreover, activating mutations of AC might participate in these disorders (145). Alternatively, enhanced cyclase activity may result from an increased expression of a particular AC isoform. Indeed, point mutations in the promoter region of the AC3-encoding gene that are associated with decreased insulin release are observed in a rat model of type 2 diabetes (146). Conversely, reduced AC activity may also contribute to pathophysiological states. For example, patients with an unusual form of pseudohypoparathyroidism have normal Gαs protein but have reduced AC activity, suggesting the presence of inactivating mutations in ACs (147).
Soluble AC
The last mammalian AC isoform to be identified was the soluble form, sAC (24). Although this unique testis-specific and soluble enzymatic activity was identified in the mid 1970s, isolation of the corresponding protein and cDNA eluded investigators for two decades (148). The enzymatic activity diverges significantly from the membrane-bound relatives in that it is unresponsive to hormones, G proteins, and forskolin. The sAC (24) is ubiquitously expressed in low amounts, but is very highly expressed in sperm cells, consistent with the role of AC activity in sperm maturation, motility, capacitation, and the acrosome reaction (149–151). Stimuli such as GTP, G proteins, and forskolin are incapable of regulating these processes. In contrast, bicarbonate and Ca2+ strongly regulate these activities, as well as increase cAMP levels and sAC activity (152). Furthermore, the concentration range of bicarbonate at which recombinant sAC is activated (EC50 ∼ 20–50 mM) is well within the range found in epididymal fluid (30, 152). Analysis of the amino acid sequence of sAC indicates some resemblance to the membrane-bound isoforms, and to cyanobacterial isoforms of AC. The protein topology is predicted to be similar, and accordingly, most of the residues responsible for catalysis are conserved. There are two splice forms resulting in 187- and 48-kDa proteins. The catalytic domain of the enzyme is located in the N-terminal region of the full-length 187-kDa form. The truncated form lacks exon 11 and results in premature termination. Messenger RNA from the truncated form is about twenty-five percent as abundant as the full-length transcript; however, the maximal activity of the truncated form is at least ten-fold greater than the full-length form in response to bicarabonate. The precise role of the C-terminal domain of the 187-kD form is unknown. The important relationship of AC and cAMP with sperm maturation and function makes sAC a very attractive potential pharmacological target. Moreover, sAC has been postulated to function as a ubiquitous metabolic sensor, similar to ACs found in cyanobacteria (153).
SUMMARY
Our understanding of the hormonal control of intracellular cAMP concentrations has come a long way since the discovery of AC, and has benefited greatly from the application of molecular genetics and structural biology. The isolation and characterization of a gene family encoding nine membrane-bound AC isoforms and one soluble isoform has increased our appreciation for the intricate complexity of the AC signaling system. Many unanswered questions still remain. For example, why is the twelve-transmembrane domain structure preserved in nine AC isoforms instead of a simpler structure like the soluble form? More directly, what is the function of the transmembrane domains? Is AC a transporter as suggested by the authors in the first paper describing the cloning of an AC (55)? Why do the similarly related nucleotide cyclases, the GCs, incorporate a more diverse structure? If cells express multiple AC isoforms, then how do they distinguish the stimulatory or inhibitory outcomes following modulation by “on-off switch” regulators, such as Gβγ and Ca2+? It is clear from a large body of literature that G protein–mediated hormonal pathways impinge on the regulation of AC activity using distinct mechanisms, and each cyclase isoform integrates this information in a specific manner. A major avenue of research will be to continue to define the regulatory repertoires of AC isoforms and to couple this with information concerning tissue and subcellular localization. Genetic knockout approaches and further structural analyses will be necessary to understand the precise physiological and biochemical roles of each AC family member.

Roger K. Sunahara, PhD, (left) is an Assistant Professor in the Department of Pharmacology at the University of Michigan Medical School.

Ron Taussig, PhD, (right) is an Assisitant Professor in the Department of Pharmacology at the University of Texas, Southwestern Medical Center, and is a member of the Alliance for Cellular Signaling. Address correspondence to either RKS or RT. E-mail sunahara{at}umich.edu; fax 734-763-4450. E-mail ron.taussig{at}utsouthwestern.edu.
- © American Society for Pharmacology and Experimental Theraputics 2002
References
- 1.↵
- 2.↵
- 3.↵
- 4.
- 5.↵
- 6.↵
- 7.
- 8.
- 9.↵
- 10.↵
- 11.
- 12.↵
- 13.↵
- 14.↵
- 15.
- 16.↵
- 17.↵
- 18.
- 19.↵
- 20.↵
- 21.↵
- 22.
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.
- 87.↵
- 88.
- 89.
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.
- 107.
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 112.↵
- 114.
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.
- 123.
- 124.
- 125.
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵

