GAF Domains: Two–Billion–Year–Old Molecular Switches that Bind Cyclic Nucleotides
- 1Department of Pharmacology
- 2Department of Biochemistry Biological Chemistry/HHMI University of Washington Seattle, WA 98195-7280
- Send correspondence to JAB. E-mail beavo{at}u.washington.edu; fax 206-685-3822.
Abstract
GAF domains represent one of the largest families of small-molecule binding units present in nature. The first mammalian GAF domains discovered were the cGMP-binding regulatory domains of several cyclic nucleotide phosphodiesterases (PDEs). The crystal structure of the PDE2A GAF domains has provided our first look at the architecture of the binding site for the second messenger cGMP. The topology of this site differs greatly from all other previously determined cyclic nucleotide binding sites. In PDE2A, cGMP binds to a well-defined pocket in one of the two GAF domains that is analogous to the ligand-binding pocket of the distantly related PAS domains of photoactive yellow protein and FixL. The consensus cGMP-binding motif suggests strongly that only certain GAF domains will bind cGMP. Although the detailed mechanism for how cGMP binding to the GAF domain regulates catalysis remains to be determined, recent data from a GAF domain–containing cAMP-stimulated adenylyl cyclase from Anabaena suggest a mechanism conserved across two billion years of evolution. Because of their unique ligand-binding topologies, the GAF domains of PDEs are likely to offer good new targets for rational drug design.
Introduction
Cyclic GMP and cyclic AMP are second messengers in many signal transduction pathways. Cyclic GMP binds directly to ion channels, activates protein kinase G (PKG), and regulates the activity of phosphodiesterases. Cyclic AMP activates protein kinase A (PKA), which, like PKG, phosphorylates a number of protein substrates. Adenylyl and guanylyl cyclases catalyze the synthesis of cyclic nucleotides, whereas 3’,5’-cyclic nucleotide phosphodiesterases (PDEs) hydrolyze these second messengers to the inactive 5’-monophosphates. Eleven different PDE families have been identified in mammalian tissues (1). Nearly all PDEs are homodimers, with monomer molecular weights ranging from 50 to 135 kDa. The PDEs are thought to provide the major mechanism for terminating cGMP and cAMP signals and thereby greatly influence the amplitude and duration of cyclic nucleotide dependent processes.
PDEs contain a C-terminal conserved catalytic domain and one or more N-terminal regulatory segments (2). Some of these regulatory domains bind cGMP. The sequences that make up these domains were originally identified in PDE2 and PDE6 (2). However, it is now known that five PDE families (PDEs 2, 5, 6, 10, and 11) contain regulatory segments composed of two of these domains. Nearly all have the domain arrangement shown in Figure 1⇓. More recently, using bioinformatic techniques, it was determined that these sequence motifs were present in many other proteins, many of which do not bind cGMP (3). These sequences were renamed as “GAF domains” to reflect this greater diversity of sequence, expression, and function. The GAF acronym comes from the names of the first three different classes of proteins identified to contain them: cGMP-specific and -regulated cyclic nucleotide phosphodiesterase, Adenylyl cyclase, and E. coli transcription factor FhlA (3).
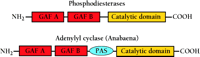
The arrangement of GAF, PAS, and catalytic domains in cyclic nucleotide phosphodiesterases and the cyanobacterial adenylyl cyclase. Nearly all of the PDEs that have been identified to date show the illustrated order of domain arrangement: two GAF domains followed by a catalytic domain. The GAF domain that is more proximal to the N terminus is designated “GAF A,” and the more distal “GAF B.” A similar arrangement is found in the cyanobacterial adenylyl cyclase, although here a PAS domain intervenes between the GAF B and the catalytic domains.
We now know that the GAF domains represent one of the largest and most widespread of the small-molecule binding domains, and are especially prominent among bacteria (4). They are distantly related to PAS domains (5), another superfamily with the same basic fold (4), and have been conserved for over two billion years of evolution, as determined by their occurrence in species from cyanobacteria to mammals. More than 1200 different GAF domain–containing proteins have now been identified in a non-redundant database (6). They are involved in many signal transduction pathways and protein regulatory and sensory systems (3, 4).
For certain PDE families, the binding of cGMP to at least one GAF domain in the regulatory segment is known to activate catalytic activity. In PDE2, cGMP bound to the GAF B domain stimulates enzyme activity more than tenfold at low substrate concentration (7–10). Cyclic GMP also has been shown to bind to PDE5 (11–13). In PDE5A, like PDE2, there are two GAF domains, and cGMP binds to the GAF A domain. The binding of cGMP to the GAF A domain allows cGMP-dependent protein kinase (PKG) to phosphorylate Ser92, just N-terminal to GAF A (14). The phosphorylation in turn increases PDE5A catalytic activity. Recent data from our laboratory and Okada et al. suggest that the binding of cGMP to GAF domains also directly activates the catalytic domains (S.D. Rybalkin, I.G. Rybalkina, M. Shimizu-Albergine, X.-B. Tang, and J.A. Beavo, unpublished observations) (15).
The photoreceptor PDE6 is expressed in rod and cone photoreceptor cells, where its catalytic activity is inhibited by its gamma subunit (Pγ), whose affinity for PDE6 is regulated by the binding of cGMP to the PDE6 GAF domain. For PDE6 there is as yet no evidence of direct cooperativity between the GAF domains and catalytic sites (16). Instead, GAF A binds the central polycationic region of Pγ and increases the affinity of the C-terminal region of Pγ for the catalytic site (17, 18).
The two recently discovered GAF-containing PDE families, PDE10 and PDE11, are poorly characterized. Their regulatory segments are homologous to those of PDE 2, 5, and 6, but definitive binding studies have not yet been reported. A preliminary study suggests that PDE10A may contain a low-affinity cGMP-binding GAF domain (19), but no allosteric effect on the catalytic site has been established.
In PDE11A, there is a possibly unique structural feature, namely, the presence of truncated GAF domains in the N-terminal region of some splice variants. A number of tissue-specific splice variants of PDE11A have been identified, although in most cases, their enzyme activities in the cell have not yet been confirmed (20–23). Nevertheless, a genomics study of the PDE11A-encoding gene shows separate promoters for both splice variants that contain an incomplete GAF A (PDE11A3) or GAF B domain (PDE11A1) (24). A preliminary study of PDE11A1 suggests that neither cAMP nor cGMP is an allosteric effector (23). However, this splice variant lacks a GAF A domain and has only an incomplete GAF B domain. Therefore, the significance of undetectable cyclic nucleotide binding is unclear. PDE11A4, which contains both GAF A and B domains, has not been reported to have cGMP or cAMP binding activity (22).
Structure of a PDE Regulatory Segment Containing GAF Domains
Recently, the overall shapes of bovine PDE6 and PDE5 have been visualized, at a resolution of 28 angstroms, from uranyl-stained electron microscopy images (25). These three-dimensional reconstructions suggest that both PDEs are dimers in which the two N-terminal GAF A regions interact with each other, but have no direct contact with the catalytic domains.
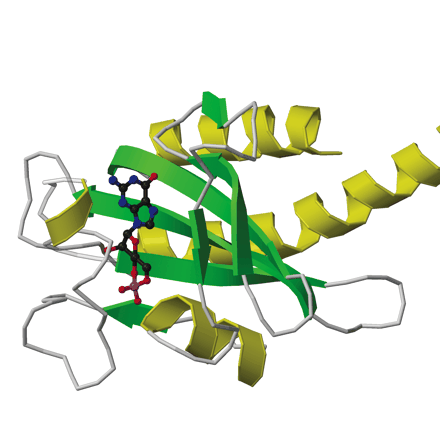
The first x-ray crystal structure of PDE GAF domains, with bound cGMP, was recently determined (Figure 2⇓) (26). A dimer of regulatory segments from mouse PDE2A, each segment containing the GAF A and B domains, was analyzed to a resolution of 2.9 angstroms. GAF A and B contain very similar folds to those seen in the structure of YKG9, a protein deduced from the yeast genome that contains a single GAF domain (27). No ligand for YKG9 has been reported; the protein does not appear to bind cGMP. In PDE2A, GAF A and B are connected by a thirty-two–residue long helix of nine turns. Unexpectedly, whereas the GAF A domains from the two regulatory segments form a dimer interface, the GAF B domains are well separated. There is also contact between the first seven turns of the connecting helices. The divergence starts after the two C386 residues, which appear (at this somewhat limited resolution) to form an intermolecular disulfide bond, although a C386S mutant structure is essentially identical to the wild-type structure. Whether this disulfide is integral to enzyme activation or positive cooperativity for substrate binding remains to be determined. Surprisingly, the two GAF A domains form an interface through a completely different set of contacts than would be predicted from the YKG9 dimer.
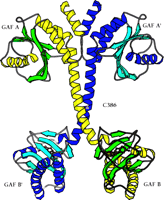
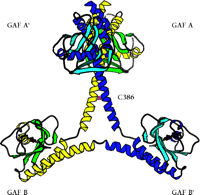
The backbone ribbon depicts the dimeric regulatory segment from PDE2A. A. The two GAF A domains interact to promote dimerization, and are adjacent to nine-turn helices that may be disulfide linked to each other at residues C386; at the C-terminal ends occur the GAF B domains that bind cGMP. B. View rotated ∼70 degrees with respect to the image in Figure 2A. Only the first seven turns of the nine-turn helices that intervene between the GAF A and GAF B sequences of each monomer are involved in dimerization. The GAF B domains are ∼65 angstroms apart. The overall dimensions of the dimerized regulatory segments are 105 by 92 by 71 angstroms. Beta sheets are colored in light blue and green; alpha helices in dark blue and yellow. Cyclic GMP is shown in red.
Although both the catalytic and GAF domains of PDEs bind cyclic nucleotides, they appear to be examples of convergent evolution, and both differ structurally from the CAP (catabolite activator protein) family of cyclic nucleotide binding proteins (28). The catalytic domain of PDE4 is a bundle of seventeen alpha helices distributed among three subdomains (29), whereas the GAF domain comprises a beta sheet packed on one side with two to four alpha helices, and on the other side with a mixture of short alpha helices and loops that form the sides of the ligand-binding pocket (27). In addition, the zinc-binding motif of the catalytic domain (30) is not present in the GAF domain.
One Molecule ofcGMP Is Bound to GAF B
Electron density was found for a single molecule of cGMP bound to the GAF B domain of PDE2A. The binding site corresponds to the PAS pocket, showing conservation between the two ancient ligand-binding motifs GAF and PAS. The structure of the GAF pocket, however, is much different from the binding pockets of other cGMP or cAMP binding proteins whose structures are known (e.g., catabolite activator protein (28), protein kinase A (31), and a nucleoside diphosphate kinase (32) for which cGMP is a competitive inhibitor). Therefore, it would seem quite feasible for selective agonists and antagonists that bind at the cGMP-binding site of the GAF domain to be developed.
The crystals of the dimeric regulatory sequences from PDE2A provide the first atomic structure of cGMP bound to a cGMP receptor (Figure 3⇓). The cGMP is completely buried, along with three bound water molecules. The most distinctive feature of the bound cGMP is that its 3’,5’-cyclic phosphate group does not contact any of the protein’s amino acid side chains; two backbone amides (i.e., I458 and Y481), however, make H-bonds to the two exocyclic oxygen atoms of the phosphate. In addition, the negatively charged phosphate group of cGMP is stabilized by the positive end of the adjacent alpha helix. The neutralization of a phosphate negative charge with a helix dipole is an important feature of the binding site. In all other experimentally determined structures that bind cAMP or cGMP, an arginine or lysine neutralizes the phosphate charge. The helix dipole, because it is derived from backbone peptide bonds, is not directly detectable on the basis of protein sequence alignments. The guanine ring and ribose moieties of the bound cGMP make a total of six polar contacts and two hydrophobic contacts with residues of the GAF domain. One residue, D439, makes both a side chain contact (to the guanine N1 nitrogen atom) and a backbone amide contact (to the C6 carbonyl group). This aspartyl residue may thus be one specificity determinant for the binding of cGMP over cAMP. Cyclic AMP has an amino group at C6, which is incompatible with the contact to the amide bond at D439, and its N1 nitrogen atom cannot engage in an H-bond with the negatively charged D439 side chain.
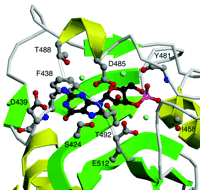
Detailed view of the cGMP-binding GAF B motif from mouse PDE2A. Shown are six side chains (D485, T488, T492, D439, S424, and E512) that make relatively close hydrogen bonds to the cGMP ligand, either directly or through one of three bound water molecules. In addition, the phenyl side chain of F438 stacks with the guanine ring, and two backbone amides (I458 and Y481) interact with the phosphate group. Beta sheets are colored green and alpha helices yellow. Cyclic GMP is shown in black. Side chains that interact with cGMP are shown in gray. Hydroxyl groups are in red and nitrogen atoms in blue. Water molecules are indicated in light blue.
As predicted from previous studies of analog-binding to PDE2, cGMP binds to the GAF domain in the anti conformation (33). Analog studies with PDE5 and PDE6 also are consistent with the prediction that cGMP will be bound in the anti conformation (12, 34). An unexpected finding is the strained boat conformation of the cyclic phosphate group. This conformation, which is normally very unfavorable, is also found for the only other crystal structure of bound cGMP, as a competitive inhibitor in the active site of nucleoside diphosphate kinase (32). It seems likely that the large number of contacts of GAF B with the bound molecule of cGMP are necessary to stabilize the strained boat conformation of the cyclic phosphate moiety and the anti conformation of the N-glycosidic linkage. Data concerning the binding of cGMP to individual PDE GAF domains have been published only for PDE5A, which shows binding to the GAF A domain (27, 35). A comparison with the PDE2A GAF B sequence indeed suggests that it is the GAF A domain of PDE5A that should bind cGMP. The poor conservation of the consensus cGMP-binding motif in the GAF B domain of PDE5A suggests that it will not provide a functional cGMP binding site. Indeed, the variety of ligands bound by PAS and GAF domains raises the intriguing possibility of unknown small-molecule ligands regulating PDEs that contain multiple GAF domains. There may well be unidentified ligands for each of the GAF domains that do not bind cGMP in PDE2, PDE5, PDE6, and possibly for the poorly characterized PDE10 and PDE11.
The structure of the PDE2A regulatory segments described in Figures 2 and 3⇑⇑ does not suggest an activation mechanism for PDE2A except for the fact that a large conformation change must take place upon binding of cGMP. The fact that the cGMP is completely buried must require that a much more open state of the binding pocket exists. However, how this conformation change in GAF B causes an activation of the catalytic domain is not yet clear and ultimately will require structural determination of the holoenzyme with and without cGMP bound. Nevertheless, it appears that the mechanism of activation extends back billions of years. In a recent paper by Kanacher et al., a cAMP-activated cyanobacterial adenylyl cyclase that contains GAF A and B domains was enzymologically characterized (36). This enzyme also has two GAF domains, like PDE2A (Figure 1⇑), but in addition contains a PAS domain between the GAF B and the cyclase domains (36). Normally, this GAF B domain selectively binds cAMP to result in activation of cAMP synthesis. When the sequence including the GAF A and B domains of rat PDE2A was swapped into Anabaena adenylyl cyclase, the chimera had a Ka with respect to cGMP of 3 μM (i.e., concentration that results in half-maximal activation), a value identical to that for rat PDE2A (36). Cyclic AMP did not stimulate this chimera. These astonishing results strongly suggest conservation of a common regulatory mechanism in an eukaryotic phosphodiesterase and a bacterial adenylyl cyclase. The intervening PAS domain in the adenylyl cyclase apparently does not block this mechanism. The mechanism by which this activation occurs is not yet understood; however, whatever the mechanism, it has been conserved for over two billion years of evolution.
Conclusion
The limited occurrence of GAF domains in mammals—along with their presence in nearly half of mammalian PDE families—makes them particularly attractive therapeutic targets for manipulation of cAMP and cGMP signaling. Homology-modeling, based on the recent crystal structure of the PDE2A GAF domains, should allow rational drug design to be initiated. Moreover, with the cGMP-binding pocket identified, it is now possible to begin addressing other questions. For example, why does the GAF B domain in PDE2Abind cGMP, whereas it is the GAF A domain that binds cGMP in PDE5A? It also appears, in the photoreceptor PDEs (i.e., PDE6A, 6B, and 6C), that GAF A better conserves the cGMP-binding motif. How can a similar regulatory mechanism operate on catalytic domains that are not always immediately adjacent to the GAF domains? What are the roles of the GAF domains that do not bind cGMP? In PDE2A, the role of the GAF A domain, which does not bind cGMP, may be to provide a dimer interface for the holoenzyme, but it is harder to predict a role of the GAF B domain of PDE5A, given that it apparently does not bind cGMP. Are there as yet undiscovered ligands for some PDE GAF domains? Such ligands could mediate a role of the GAF-PDEs in crosstalk with other signaling networks. Clearly, the new insights into the structure of the GAF motifs of PDE2A will be important as investigators address these and other questions.
GAF DOMAINS AS DRUG TARGETS
The SMART domain database contains (as of 8/5/02) approximately 1200 GAF domains, of which only about fifty-five pertain to mammals. In humans, nearly all of the GAF domains identified are in the PDEs. Although additional GAF domains may be discovered with further annotation of the human genome, the small percentage that have already been established suggest that synthesis of specific regulatory molecules that bind to the GAF domains of mammalian PDEs may be possible.
Rational drug design typically has been used to design competitive inhibitors of active sites. Some early examples of such design are the HIV protease inhibitors, Crixivan and Saquinavir. In addition to the rational synthesis of active-site ligands, however, small molecules could also be designed to target regulatory domains. Two types of functional ligands could be designed to bind GAF domains: agonists and antagonists. An antagonist might bind to the open (i.e. cGMP-free) conformation of the GAF domain and stabilize it in that state, blocking the natural ligand from binding and closing the GAF domain. The result would be an enzyme left in the inactive state. A GAF agonist, on the other hand, might bind more tightly to the closed conformation than the natural ligand, permanently activating the enzyme.
Both PDE2A and PDE5A could be attractive targets for GAF-binding agonists or antagonists. The activation of PDE2A by atrial natriuretic peptide, a hypotensive hormone, decreases cAMP levels in the adrenal cortex, thereby decreasing production of aldosterone and retention of salt by the kidney (37). An agonist that would bind the GAF B domain of PDE2A, for which a crystal structure is available, could similarly be used to fine-tune treatment of high blood pressure.
For PDE2A, a GAF-binding agonist would not necessarily have to resemble cGMP. No structure of the unbound GAF B is available, but presumably the domain would go through an open conformation before binding cGMP. Such an open conformation might be an actual broadening of the ligand pocket. Interestingly, an alpha helix that lies over the cGMP molecule in the closed conformation makes distinct contacts with the ligand through four amino acid side chains; in the absence of these four contacts—that is, in the unbound state—it is possible that this protein sequence is disordered. A GAF-binding agonist probably would preserve some feature of cGMP, such as the negative charge where the phosphate group is pointed to the adjacent helix (see Figure 3⇑). The three bound water molecules offer some added flexibility for drug design, because they could be displaced if necessary.
Since it has recently been found that cGMP binding to the GAF A domain of PDE5 will also stimulate catalysis, antagonists of this enzyme would also be of interest. Importantly, this site would be expected to have different binding requirements than the active site. Active-site inhibitors like sildenafil (Viagra®) have proved very useful for increasing cGMP in vascular smooth muscle and for treatment of erectile dysfunction. By blocking the binding of cGMP to GAF A, no phosphorylation and activation of PDE5 by protein kinase G could occur, thereby possibly potentiating inhibition by sildenafil. On the other hand, a GAF A domain–binding agonist might activate the enzyme at lower concentrations of cGMP and thereby blunt a large increase in cGMP levels in response to pathologic stimuli. Such activation might for example be useful for treatment of ischemic injury after stroke, because high levels of cGMP have been associated with this pathology (38). It would seem very difficult, on the other hand, to design an agonist that would occupy the catalytic site and yet allow for the binding of substrate. Finally, if ligands for the GAF domains that do not bind cGMP can be found, these domains may also prove useful drug targets.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Sergio Martinez, PhD, is a postdoctoral investigator (top) in the Beavo and Hol labs. Joe Beavo, PhD, is Professor of Pharmacology at the University of Washington in Seattle, and a member of the National Academy of Sciences (center). Wim Hol, PhD, is Professor of Biochemistry in Seattle and a Howard Hughes Medical Institute investigator (bottom).

