Apolipoprotein E, Amyloid, and Alzheimer Disease
- 1Neuroscience Discovery Research, Lilly Research Laboratories Indianapolis, IN 46285
- 2Washington University School of Medicine, 660 S. Euclid Ave. St. Louis, MO 63110
- Address correspondence to SMP. E-mail Paul_Steven_M{at}lilly.com; fax 317-277-1125.
Abstract
The alleles for apolipoprotein E (apoE) represent important genetic risk factors for the most common late-onset forms of Alzheimer disease (AD), with the ε 4 and ε 2 alleles increasing and decreasing the risk for developing AD, respectively. ApoE, a 34-kDa lipid transport protein, is predominantly expressed in the liver, but is also expressed in brain by microglia and astrocytes. Studies utilizing mouse models that mimic the neuropathology of AD have demonstrated an apoE isoform–dependent effect on amyloid-β peptide (Aβ ) deposition, fibrillization, and neuritic plaque formation. Taken together, these data support an important (and isoform-dependent) role for apoE in the pathogenesis of AD, most likely by altering Aβ clearance and/or metabolism. Further elucidation of the exact cellular and molecular events mediating apoE isoform–dependent amyloid deposition could lead to novel therapeutic strategies for preventing or treating AD.
Introduction
Alzheimer Disease (AD) is now the eighth leading cause of death in the U.S. (1) . This neurodegenerative disorder, characterized by progressive loss of cognitive abilities leading to frank dementia, is invariably fatal and often results in institutionalization during the latter stages of the disease. As the population of the developed countries—which overall enjoy reduced morbidity and mortality for most diseases—ages, and as life expectancies increase, AD will become an ever-increasing and pandemic public health burden. Currently available therapies, for the most part, target a deficiency in the cholinergic neurotransmitter system, a neurochemical system largely thought to be involved in short term memory, and which is severely affected in AD (2) . Currently available medications, however, offer only palliative symptomatic relief. Many new strategies yet to be tested in the clinic will, hopefully, offer not only symptomatic treatment of the cognitive deficits, but more importantly will prevent or slow disease progression. A near-term test of the prevailing hypothesis of AD pathogenesis, “the amyloid hypothesis,” centers around disrupting the neuropathological events leading to the extracellular deposition of the amyloid β -peptide (Aβ ) and subsequent formation of neurofibrillary tangles that are diagnostic of AD at autopsy (3) . Although still unproven, the amyloid hypothesis is supported by ample genetic epidemiological data, especially for the rare early-onset (<65 years of age) and familial forms of the disease. However, over the past few years, considerable data supporting the amyloid hypothesis has also been derived from studies of apolipoprotein E (apoE), an important risk factor for late-onset sporadic AD (4) . Here, we review the data supporting an important role for apoE in determining brain amyloid burden and the formation of neuritic plaques, neuropathological hallmarks of AD.
Genetic Evidence Supporting the Amyloid Hypothesis of Familial AD
Perhaps the best evidence for the amyloid hypothesis comes from studies of the rare autosomal dominant forms of familial AD, where mutations in genes encoding either the amyloid precursor protein (APP), located on chromosome 21, or the presenilins (PS1, PS2) located on chromosome 1 and 14, respectively, have been shown to segregate with disease. Although relatively rare (comprising <5% of all AD cases), mutations within the APP or PS genes result in a neuropathological and clinical phenotype that is indistinguishable from the more common late-onset sporadic forms of AD (5–9) . Significantly, the majority of the missense mutations in APP that are linked to familial AD have been reported to alter the generation of the amyloid-β peptide (Aβ ). For example, the “Swedish” APP mutation (i.e., the double mutation, K670N/M671L, just N-terminal to the site at which β -secretase cleaves APP) results in enhanced production of two forms of Aβ : Aβ 40 and Aβ 42 (9) . Furthermore, plasma Aβ 42 levels from FAD patients with PS1 gene mutations are elevated, suggesting increased generation of what is thought to be the more amyloidogenic form of the Aβ peptide, which results from endoproteolytic cleavage of APP by β and γ secretases, respectively (8–11) . Additionally, trisomy of chromosome 21 (Down syndrome) results in a triplication of the APP-encoding gene (increased gene dose), increased Aβ synthesis, and an early-onset form of AD (12) . Taken together, these data support the hypothesis that familial AD results from the overproduction of the Aβ peptide(s), and further suggests that late-onset AD could occur through similar mechanism(s). Nevertheless, for the vast majority of patients with AD (especially the more common late-onset forms), there is no evidence for an increase in Aβ synthesis.
APOE Alleles: Major Genetic Risk Factors for Late-Onset Alzheimer Disease
Polymorphisms in other genes (which are distinct from either the APP or PS loci), and which do not appear to have any overt effect on the generation of the Aβ peptides, have also been identified as altering (increasing or decreasing) the risk of developing AD (13–15) . The best characterized of the late-onset susceptibility genes for AD, the ε 4 allele of the APOE , is now widely regarded as an important risk factor for AD, and is present in thirty to fifty percent of patients who develop late-onset AD (15) .
ApoE is a circulating 34-kDa secretory protein, synthesized primarily in the liver, and functions in the periphery as a mediator of lipoprotein metabolism through the binding of apoE-containing plasma lipoprotein particles to members of the low density lipoprotein (LDL) superfamily of receptors. Within the central nervous system (CNS), apoE is synthesized and secreted primarily by astrocytes and microglia, and its importance is underscored by the absence of most other plasma apolipoproteins in brain (16) . Additionally, brain apoE is believed to play a role in the redistribution of lipid and cholesterol during membrane repair and synaptic plasticity as well as in the transport of apoE-containing lipoproteins in the cerebrospinal fluid (17,18) .
In humans, there are three common APOE alleles: ε 2, ε 3, and ε 4. Remarkably, the three apoE isoforms, which differ by only one or two amino acids (E2 Cys112 Cys158 ; E3 Cys112 Arg158 ; E4Arg112 Arg158 ), confer at least a twentyfold difference in risk for developing AD (E4 > E 3> E 2) (Table 1⇓). The critical observation linking the ε 4 allele to late-onset AD occurred when investigating the APOE allele frequency (ε 2, ε 3, and ε 4) in a cohort of patients with late onset AD and healthy age-matched control subjects (19) . Individuals who inherit one or two copies of the ε 4 allele have a greater risk (two- to tenfold, respectively) as well as an earlier age of onset for developing AD (15) . The increased frequency of the ε 4 allele in AD patients, compared to age-matched healthy controls, has now been widely reproduced for both late-onset as well as sporadic cohorts of AD patients, in numerous genetic epidemiological studies from a variety of populations of diverse ethnic origin (15, 16) . Even in Japan, where the ε 4 allele frequency is relatively low, its association with AD has been established (15, 20) . By contrast, inheritance of the ε 2 allele appears to reduce the risk of developing AD, even when combined with the ε 4 allele or in patients with Down syndrome (21, 22) . Moreover, the presence of the ε 2 allele appears to preserve cognitive function in Down syndrome patients as they age (22) . It should be emphasized, however, that the ε 4 allele of APOE does not confer an absolute predisposition to develop AD, as reflected by centenarians who are ε 4 homozygotes and are disease free (23) . Thus, the ε 4 allele is a true genetic risk factor, most likely interacting with other genetic and nongenetic factors to facilitate AD.
ApoE Isoforms in the Human Population
The amino acid differences among the three common apoE isoforms occur in critical regions of the amino terminal portion of the molecule which influences the lipoprotein binding preference (located in the carboxy terminal portion of the protein) in an isoform-dependent fashion (16) . Very low-density lipoproteins are the preferred binding partners for the E4 isoform of apoE, whereas high-density lipoproteins are preferred by the E3 isoform. These critical domain interactions (which specify the phenotype of the lipid particles) are thought to underlie the predisposition of ε 4 carriers to hypercholesterolemia. To date, very little is known, however, about the exact role of apoE particles in the CNS; however, it is likely that particle phenotype and isoform preference contributes, in some way, to the ε 4 isoform-dependent susceptibility for developing AD.
Apolipoprotein E and Amyloid: Circumstantial Evidence
It appears that both cholinergic deafferentation (neurodegeneration) of the hippocampus as well as the severity of Aβ deposition and amyloid burden are greater in ε 4 carriers (19, 24) who die with AD. Neuropathological examination of a large cohort of late-onset AD patients with one or two ε 4 alleles revealed significantly more Aβ deposition in both the cerebral cortex as well as cerebral vessels. Moreover, the number and size of Aβ deposits has been reported to be greater in ε 4 carriers (25) . In a separate study, Hyman and colleagues mathematically modeled senile plaque distribution in AD patients who were either ε 4 carriers or Down syndrome patients, and based on their model, they hypothesized that the increased number of senile plaques in ε 4 carriers was the result of an accelerated rate of Aβ deposition (due to an increased probability of senile plaque initiation) in brain (26) . By contrast, those patients with AD due to Down syndrome (increased APP gene dose) had larger plaques, suggesting that the increase in brain amyloid burden in these two subgroups of AD are due to different molecular mechanisms. Interestingly, the association between the apoE (ε 4) genotype and brain amyloid burden has now been extended to a number of other brain disorders as well as normal aging. Increased amyloid burden has been reported for ε 4 carriers who have Down syndrome, amyloid angiopathy, acute head trauma, and chronic brain injury (27–31) . Moreover, the prognosis following ischemic injury or stroke is poorer for ε 4 carriers when compared to the other apoE alleles (32, 33) . More recently, polymorphisms within the apoE gene promoter have been found to correlate with an increased incidence of AD, suggesting that a critical level of apoE protein (perhaps independent of the isoform expressed) may be necessary for normal brain function and (or) Aβ /amyloid formation (34, 35) . Some investigators have reported an increase in apoE mRNA or protein in the brain and cerebrospinal fluid of AD patients compared to controls, further suggesting that apoE expression may increase, perhaps in response to aging or the deposition of certain forms of Aβ in brain (36, 37) . Additionally, the C- terminal portion of apoE not only forms amyloid-like fibrils when incubated alone in vitro, but also copurifies with Aβ when extracted from neuritic plaques. Because the C-terminal region of apoE is identical among the different isoforms, and apoE has been shown to be associated with systemic amyloidosis, also in an isoform-independent fashion, apoE has been termed a “pathological chaperone,” denoting its importance in facilitating amyloid formation in brain parenchyma (38, 39) .
Apolipoprotein E and Amyloid: Direct Evidence
Indirect evidence suggests that the interaction between apoE and Aβ may in some way be critical to the isoform-dependent effects of the ε 4 allele on brain amyloid burden in AD as well as other brain disorders. To directly investigate the role of apoE on amyloid deposition, our group has utilized transgenic mice overexpressing a mutant form of APP (i.e., APP V717F ) that is associated with familial AD. These mice are also commonly referred to as “PDAPP mice” because the transgene is expressed behind the promoter for the platelet derived growth factor β subunit (40) . APP V717F transgenic mice represent a well-characterized rodent model of AD, manifesting both age- and region-dependent deposition of Aβ and amyloid (i.e., thioflavine S-fluorescent or congophilic deposits), neuritic plaques, and an associated astrogliosis, microgliosis, and synaptic loss. By crossing APP V717F transgenic mice to mice lacking apoE (apoE knockout mice), we generated mice with zero, one, or two copies of the gene for murine apoE (41–44) . While mice lacking apoE still develop diffuse, non-fibrillar Aβ deposits, they have no fibrillar amyloid deposits when assessed at young ages, and markedly reduced amyloid burden was observed in very old mice, even when the brain level of Aβ itself approaches that found in APPV717F transgenic mice with a full complement of murine apoE (Figure 1⇓). Importantly, we observed an APOE gene dose-dependent effect on brain amyloid burden in that only half the amount of amyloid ever accrues in mice with only a single copy of the gene (Figure 2⇓). Thus, apoE expression, although not absolutely essential, greatly facilitates amyloid formation in APPV717F transgenic mice. In a subsequent study, we quantified the number of neuritic plaques in the hippocampus and cerebral cortex of APPV717F transgenic mice with and without apoE (43) . Neuritic plaques, defined as Aβ deposits associated with marked neuritic dystrophy, colocalized with virtually all amyloid deposits, measured by thioflavine S-fluorescence or Congo red birefrigence. The number of neuritic plaques was also found to be apoE-dependent, such that by fifteen months of age, APP V717F transgenic mice expressing murine apoE had nearly twenty-five times more neuritic plaques in the hippocampus than mice lacking apoE (Figure 3⇓).


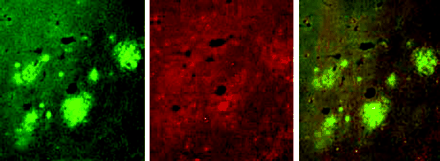
Lack of murine apoE dramatically reduces the amount of Aβ/amyloid burden even in very old transgenic mice. Numerous thioflavine S-fluorescent (A) as well as Aβ immunoreactive deposits (C) are evident in both the cortex and hippocampus of transgenic mice that express the V717F mutant of the amyloid precursor protein as well as apoE, but is dramatically reduced when apoE is absent (B, D). Adapted from (42).
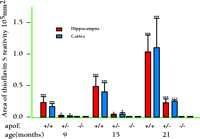
Amyloid plaques are quantitatively related to the dosage of the Apoe gene. The brain area occupied by thioflavine S-fluorescence was quantified in both hippocampus and cortex of APPV717F transgenic mice with zero, one, or two copies of the Apoe gene. Note the nearly complete absence of amyloid (thioflavine S-fluorescent deposits) in mice lacking murine apoE. Mice with only one copy of the murine gene have less than half the amount of amyloid (thioflavine S-fluorescent deposits) relative to mice with two copies of the Apoe gene, even though the amount of Aβ immunoreactiviy is nearly identical to that of mice with two copies of the gene. (Data not shown.) Adapted from (42).
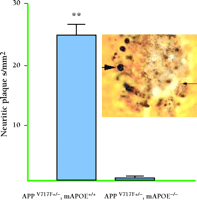
Neuritic plaques (as determined by the de Olmos silver stain) in APPV717Ftransgenic mice with and without apoE (at fifteen months of age). APPV717F transgenic mice that express apoE have nearly twenty-five times more neuritic plaques compared to mice without apoE. Inset: Large arrow points to a large, distended, dystrophic neurite in a plaque, and small arrow points to fine dystrophic neurites. Adapted from (43).
Similar brain Aβ /amyloid deposition occurs in a variety of transgenic models that overexpress APP (e.g., Tg2576, APP23, and APPV717F , as well as double mutants overexpressing mutant presenilin genes). All of these transgenic animals are characterized by similar anatomical deposition of Aβ and amyloid, despite differences in promoters, transgene constructs, and genetic background. Importantly, the lack of apoE in Tg2576 mice also resulted in a dramatic reduction in amyloid deposition in the brain parenchyma as well as in the cerebrovasculature. Similar to results in APPV717F transgenic mice lacking apoE, little neuritic degeneration was found in Tg2576 mice that lacked apoE (45) .
To explore the potential isoform-dependent effects of apoE on amyloid deposition in vivo, we generated mice expressing human apoE3 or apoE4 (but devoid of murine apoE) under the control of the promoter for the gene that encodes glial fibrillary acidic protein (GFAP) (Figure 4⇓). In transgenic mice older than fifteen months that express either human apoE3 or apoE4, we observed an isoform-dependent effect on amyloid burden; specifically, apoE4-expressing mice had approximately ten times more amyloid than apo E3-expressing mice (43) . Similarly, the number of neuritic plaques in the apoE4-expressing mice was approximately threefold greater than the number of neuritic plaques present in age-matched E3-expressing mice (43) .

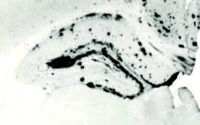
Expression of human apoE4 by astrocytes results in an isoform-dependent effect on Aβ deposition. Expression of human apoE4 (B) and apoE3 (C) recapitulates the anatomical distribution of Aβ deposition seen in APPV717F transgenic mice expressing murine apoE (A). Significantly more Aβ deposition, however, is associated with murine apoE (murine apoE>>>human E4>>human E3). Adapted from (43).
Because apoE immunoreactivity colocalizes with nearly all neuritic plaques in AD-affected brains, we used immunohistochemistry to examine the degree to which apoE is associated with Aβ and amyloid deposits in APPV717F transgenic mice (42) . Similar to what has been reported in postmortem human brain tissue from AD patients (46) , we found that apoE was associated with only a subset of Aβ deposits (approximately ten to fifty percent, depending on brain region) but that virtually all amyloid deposits, as detected by immunostaining, were associated with apoE. In a separate study, we measured both soluble and insoluble apoE in brain extracts from APPV717F transgenic mice at early or late stages of plaque development (47) . We found that the levels of insoluble apoE were greatly elevated in the hippocampus and cortex of aged transgenic mice, but were unaltered in young APPV717F or wild-type mice. Significantly, we found a positive correlation between the levels of apoE and Aβ in the insoluble fraction in both the hippocampus and cortex (i.e., regions displaying amyloid deposits), but not in the cerebellum (i.e., a region that does not show amyloid deposition; 47 ) (Figure 5⇓). Finally, we found an approximate threefold increase in Apoe mRNA in the hippocampus of older (plaque-bearing) APPV717F transgenic mice, compared to young (plaque free) transgenic mice, suggesting that apoE expression is increased, perhaps as a compensatory inflammatory response to the sequestered insoluble apoE. Taken together, these findings strongly suggest that a direct physical interaction between apoE and Aβ facilitates fibrillization as well as amyloid and neuritic plaque formation. This latter observation also suggests the possibility that the effects of apoE on Aβ deposition, aggregation, and fibrillization may be age-dependent. Thus, in an age- and isoform-dependent manner, apoE may facilitate Aβ clearance (E2>E3>E4) and/or Aβ fibrillization and subsequent amyloid formation (E4>>E3>E2).
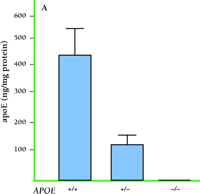
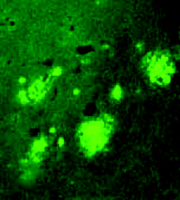
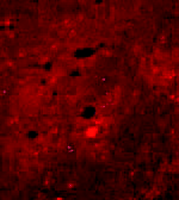
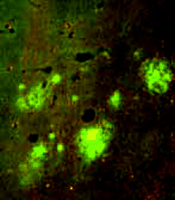
Expression of apoE and its association with Aβ deposits in transgenic mice. A. Levels of apoE in APPV717F transgenic brain homogenates correspond to Apoe gene dosage. B. Staining for apoE (green) and Aβ (red) shows that apoE colocalizes with most Aβ -immunoreactive deposits (bottom panel). Adapted from (47).
Apolipoprotein E and Aβ Deposition
The lack of apoE in brain parenchyma also had profound effects on the neuroanatomical distribution of the depositing Aβ peptides in APPV717F transgenic mice. Significantly fewer Aβ deposits were observed in the cerebral cortex and outer molecular layer of the dentate gyrus of the hippocampus, while significantly more Aβ immunoreactivity was apparent in the hilus of the dentate gyrus, including areas of the stratum oriens and radiatum (41–43, 48) . Moreover, these dramatic effects on the anatomical distribution of Aβ immunoreactivity occurred independent of any effects on the relative levels of APP, soluble Aβ or LRP, an apoE receptor (48) . The dramatic anatomical redistribution of Aβ deposition in the hippocampus of APPV717F transgenic mice in the absence of apoE was not observed in Tg2576 mice lacking apoE, but suggests a potentially novel effect of apoE on the processing of APP in APPV717F transgenic mice. In support of this hypothesis, we have recently demonstrated that, in the absence of apoE, the accumulation of anti-Aβ immunoreactive multifocal aggregates in the dentate gyrus results, at least in part, from the accumulation of β -cleaved C-terminal fragments of APP (49) .
To investigate whether expression of human apoE3 or -E4 affected the deposition of the Aβ peptide, we quantified Aβ immunoreactivity in APPV1717F transgenic mice expressing either isoform. Similar to the effects on amyloid deposition (see above), astrocytic expression of human apoE3 or -E4 resulted in an isoform-dependent effect on Aβ deposition (43) . By fifteen months of age, APPV717F transgenic mice expressing human apoE4 had significantly more Aβ mmunoreactivity in the hippocampus, relative to age-matched mice expressing human apoE3. Importantly, the expression of human apoE3 or -E4 resulted in Aβ deposition in the hippocampus, which recapitulates the anatomical pattern seen in APPV717F transgenic mice expressing murine apoE (43) (Figure 4⇑). Whereas these results suggest that, at older ages, an isoform-dependent effect (E4>E3) occurs on both Aβ as well as amyloid deposition, we also observed that human apoE expression (regardless of isoform) delays the onset of Aβ deposition by several months, relative to murine apoE (44) . This effect was rather dramatic in that the amount of Aβ deposited in nine-month-old transgenic mice expressing either E3 or E4 was significantly less, even when compared to age matched transgenic mice lacking apoE. More recently, we have extended this observation to a larger and older cohort of transgenic mice expressing human apoE2, -E3, or -E4 (50) . Remarkably, even at eighteen months of age, transgenic mice expressing the human apoE isoforms have greatly reduced brain Aβ /amyloid burden compared to transgenic mice expressing murine apoE. However, the amount of amyloid deposition we observed was isoform-dependent and correlated with neuritic plaque pathology (E4>>E3>E2). Interestingly, the neuroanatomical distribution (especially in the hippocampus) of Aβ deposition in transgenic mice expressing human apoE2 was very similar to the pattern seen in apoE-deficient transgenic mice. These observations suggest that a critical level and/or form of the Aβ peptide and/or apoE must exist in brain parenchyma prior to Aβ /amyloid deposition, which further supports our working hypothesis that apoE expression in brain may promote both clearance (E2/E3>>E4) as well as fibrillogenesis (E4>>E3/E2) of the Aβ peptide.
Apolipoprotein E and Aβ Clearance: Possible Mechanisms
Given the rather robust effects of apoE expression on Aβ deposition in APPV717F transgenic mice, and the well-recognized ability of apoE to bind to and form stable complexes with Aβ (51) , it seems reasonable to postulate that apoE may, in an isoform-dependent manner, alter the clearance or metabolism of Aβ in brain. Conceivably, therefore, isoform-dependent differences in the binding of apoE to Aβ and subsequent binding of the apoE–Aβ complex to one or more apoE receptors could account for observed isoform-dependent effects of apoE on Aβ deposition. In fact, Koo and colleagues have recently reported that another Aβ binding protein, α 2 macroglobulin (α 2M), can sequester and clear Aβ in vitro via the LDL-like receptor (LRP), one of the important apoE receptors expressed in brain (52, 53) . If apoE serves a similar clearance function for extracellular Aβ , an isoform-dependent difference in clearance (E2/E3>>E4) could contribute to extracellular Aβ deposition, aggregation, and fibrillization, especially as one ages. Moreover, Russo and colleagues have found that apoE–Aβ complexes isolated from the brains of AD patients are less stable and more sensitive to proteinase K digestion in vitro than complexes isolated from age-matched (non-AD) control brains (54) . Here again, one might postulate that apoE4 may form a less stable complex with Aβ than do apoE3 and apoE2, thereby rendering Aβ less sensitive to degradation by endogenous proteases and more likely, therfore, to aggregate and deposit in brain parenchyma. Whereas isoform-dependent differences in Aβ binding, clearance (by one or more apoE receptors), or metabolism have not as yet been unequivocally demonstrated in vivo, such differences are, in our view, likely to explain the observed isoform-dependent differences in Aβ deposition and amyloid burden.
Very recently, evidence has also accumulated to indicate that centrally derived Aβ may be cleared from the CNS via transport/efflux to the peripheral circulation (55) . Thus, Aβ binding proteins such as apoE and apolipoprotein J, as well as Aβ /plaque deposition itself, may influence Aβ clearance from brain (56, 57) . In support of this notion, we have found that plasma Aβ levels in mice lacking apoE are significantly higher when compared to mice with a complete complement of mouse apoE (K.R. Bales and S.M. Paul, unpublished observations). Additionally, the level of plasma Aβ decreases in older (plaque-bearing) transgenic mice that express apoE, relative to younger (non-plaque-bearing) aged transgenic mice with a similar genetic background (K.R. Bales and S.M. Paul, unpublished observations). Although apoE expressed within the brain may alter the clearance of Aβ out of the brain, the rate of transport of radiolabelled Aβ when injected into the ventricle is apparently unaffected by apoE (58) . Nonetheless, a careful assessment of the effects of various apoE isoforms on endogenous Aβ metabolism and clearance from brain will likely prove informative.
Apolipoprotein E and Neurofibrillary Tangle Formation
Could there also be an apoE isoform–dependent effect on the phosphorylation of the microtubule associated protein, tau, and the formation of paired helical filaments, the cytoskeletal hallmark of neurodegeneration found in AD brain? Several studies have reported an apoE4-dependent increase in phosphorylated tau, suggesting an isoform-dependent effect on neurofibrillary tangle formation reminiscent of the isoform-enhancing effect on Aβ and amyloid plaque burden (59–61) . Although the exact mechanisms by which apoE might alter the phosphorylation of tau is unclear, several possibilities exist, including a direct physical interaction of apoE and tau (62) . Whether or not apoE is expressed in neurons, is controversial; however, apoE is clearly “taken up” by neurons and specific colocalization of apoE protein within neurons has been reported (63) . It seems reasonable to speculate, therefore, that the selective uptake of apoE, by one or more neuronal apoE receptors may, in turn, result in specific, isoform-dependent effects of apoE on tau phosphorylation. In fact, overexpression of apoE4 in neurons (utilizing the Thy 1 promoter) in transgenic mice has been reported to dramatically increase tau phosphorylation (61) . Additionally, it is also plausible that different forms of Aβ that may be associated with specific isoforms of apoE-containing particles, and that again, when internalized by neurons, may result in intracellular accumulation/ aggregation of various Aβ peptide(s) leading to hyperphosphorylation of tau and subsequent neurodegeneration. Finally, the kinases/phosphatases responsible for the phosphorylation/dephosphorylation of tau may be sensitive to a specific isoform of apoE and/or type of Aβ peptide associated with a particular apoE-laden particle.
Neuroinflammation, Glial Activation and Apolipoprotein E
Neuroinflammation, resulting in or resulting from, Aβ deposition in AD-affected brain is widely believed to contribute to the pathogenesis of AD (64) . Although activation of the complement cascade, upregulation of acute-phase proteins, cytokines, chemokines, and their receptors appear in conjunction with brain amyloid deposits, a direct causal link between these inflammatory stigmata and Aβ deposition and amyloid formation and the progression of AD remains elusive. Could apoE contribute to the neuroinflammatory cascade in a pathological fashion? Conversely, could apoE, in an isoform-dependent fashion, be responsible for beneficial “inflammatory” responses that may occur during the initial or even later phases of AD?
In an attempt to address this question, we injected lipopolysaccharide (LPS), a gram-negative bacterial endotoxin, directly into the ventricle of APPV717F transgenic mice with or without apoE (65) . A significant increase in amyloid (thioflavine S-fluorescent positive) deposits was apparent in young (three-month-old) transgenic mice following LPS administration. Moreover, apoE mRNA, as determined by in situ hybridization, was increased by LPS treatment in these same mice. Significantly, no amyloid deposits were apparent in transgenic mice devoid of apoE. Interestingly, an increase in the mortality of apoE knockout mice following LPS administration suggests that apoE in the periphery (and perhaps brain) may mitigate, either directly or indirectly, the “toxic” inflammatory effects of LPS exposure.
In vitro, glia have been reported to respond to exogenously added cytokines as well as the Aβ peptide itself by increasing levels of apoE protein and corresponding mRNA (66, 67) . Pretreatment of BV2 cells (a murine microglia cell line) with salicylic acid (which inhibits NF-κ B activation) or antisense oligonucleotides to the p65 subunit of NF-κ B attenuates Aβ -induced apoE release (66) . Because the NF-κ B/Rel family of proteins are well-characterized mediators of the peripheral inflammatory response, our results suggest that Aβ as well as other cytokines (e.g., IL-1β ) that are upregulated in AD-affected brain, may promote and/or initiate an inflammatory cascade through a similar pathway. Furthermore, drugs including some nonsteroidal anti-inflammatory agents may be therapeutically efficacious because of their ability to inhibit NF-κ B activation and subsequent Aβ /cytokine -induced glial activation.
ApoE has also been reported to have anti-inflammatory effects on a variety of inflammatory cell types, and therefore, may play a critical role in attenuating the CNS effects of inflammatory stimuli (e.g., Aβ itself or the cytokines IL1-β and TGF-β ) (68) . Thus, isoform-dependent differences in the anti-inflammatory properties of apoE may contribute to AD pathogenesis. In support of this hypothesis, Colton and colleagues have recently reported isoform-dependent differences in the relative levels of nitric oxide (NO) produced by microglia from ε 2, ε 3, and ε 4 transgenic mice (69) . More NO was evident in apoE4-expressing microglial cultures relative to cultures expressing murine apoE or any of the other human apoE isoforms. These data suggest that apoE4 may be a less effective anti-inflammatory isoform and may therefore contribute to an acceleration of AD. Thus, certain kinds of anti-inflammatory agents may be effective in preventing or slowing disease progression in ε 4 carriers who have or will develop AD.
Apolipoprotein E: Diverse Functions in the CNS
In cultured neurons, apoE has been shown to induce neurite outgrowth and to function as an endogenous antioxidant (70–72) . These effects were also reported to be isoform-dependent (apoE2/E3>>apoE4). More recently, apoE receptors, which are highly expressed on neurons, have been shown to be directly involved with transmitting positional cues for migrating populations of neurons (73) . For instance, binding of reelin, the extracellular ApoE receptor-2 (ApoER2) ligand, is responsible for directing Disabled-1, an intracellular adapter protein, to the cytoplasmic tail of ApoER2 (74) . Similarly, it has been shown that LRP is directly involved in synaptic transmission in the adult brain inasmuch as ligand binding enhances the activity of protein kinase A, a pivotal kinase involved in long-term potentiation (75) . Moreover, binding of α 2M, another ligand of the LRP receptor, results in site-directed NMDA-specific Ca+2 influx in neurons (76) . Similar to the LDL receptor gene family that subserves complex signaling as well as endocytic functions, apoE is well positioned to modulate multiple biological effects, both pre- and postsynaptically, the potential diversity of which may be amplified by the range of apoE isoforms expressed (Figure 6⇓). Determining whether or not any of these diverse biological functions of apoE contribute to its role in AD pathogenesis will require further study.
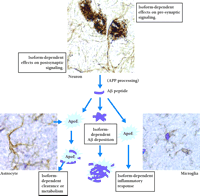
Potential sites for apoE isoform–dependent effects.
Summary
The APOE alleles represent important genetic risk factors for the most common late-onset form(s) of AD, with the ε 4 and ε 2 alleles increasing and decreasing the risk for developing AD, respectively. Both indirect as well as direct evidence support a critical role for apoE in the deposition and fibrillization of Aβ peptides to form amyloid deposits and neuritic plaques, two key neuropathological features of AD. Using various strains of transgenic mice overexpressing a mutated APP transgene (i.e., APPV717F transgenic mice), together with various combinations of the murine and human apoE isoforms, we have demonstrated isoform-dependent effects of apoE on Aβ deposition and amyloid as well as neuritic plaque formation. Interestingly, expression of human apoE initially reduces Aβ deposition in APPV717F transgenic mice, compared to mice devoid of apoE or expressing murine apoE. However, as these mice age, apoE isoform–dependent effects on Aβ deposition, amyloid and neuritic plaque formation are observed. Our findings have established an important role for apoE in facilitating brain amyloid burden and in determining the number of neuritic plaques formed in aged APPV717F transgenic mice. We postulate that apoE may play a similar role in the pathogenesis of AD. The cellular mechanisms underlying the effects of apoE on amyloid deposition and neuritic plaque formation are still poorly understood, but most likely involve a direct interaction between apoE and Aβ that alters the metabolism or clearance of Aβ , or both. Because apoE is expressed primarily in glia (both astrocytes and microglia), and because glial activation is a prominent feature of AD (especially around Aβ deposits, including neuritic plaques), it is possible that apoE may, in part, mediate the postulated effects of “neuroinflammation” in AD pathogenesis. By inference, drugs that reduce glial activation and apoE (especially apoE4) expression may inhibit amyloid deposition and neuritic plaque formation. Finally, although our data strongly implicate apoE as a “pathological chaperone” in altering Aβ clearance and faciliting brain amyloid deposition, other neurobiological effects of apoE may also contribute to AD pathogenesis. Nevertheless, the data on apoE reviewed herein provide additional and strong support for the amyloid hypothesis of AD, especially for the more common late-onset form(s) of the disease.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Kelly R. Bales (seated, foreground) is Senior Scientist in Neuroscience Discovery Research at Eli Lilly & Co., and Steven M. Paul, MD , is Group Vice President, Therapeutic Area Discovery Research at Lilly. David M. Holtzman, MD , is the Charlotte and Paul Hagemann Professor of Neurology at Washington University School of Medicine. Ronald B. DeMattos, PhD (far left), and Jean-Cosme Dodart, PhD (standing, rear), are postdoctoral fellows in Neuroscience Discovery Research at Eli Lilly & Co.

