Two Birds with One Stone: Novel Glucokinase Activator Stimulates Glucose-Induced Pancreatic Insulin Secretion and Augments Hepatic Glucose Metabolism
- 1Department of Pharmacology, German Institute of Human Nutrition, 14558 Potsdam-Rehbrücke, Germany;
- 2Department of Psychiatry, Obesity Research Center, University of Cincinnati, Cincinnati, OH, 45221;
- 3Diabetes Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA 20892
The insulin-insensitive form of diabetes, Type 2 diabetes mellitus (T2DM)—most frequently arises as a consequence of obesity—represents approximately 95% of the overall incidence of diabetes, afflicting an estimated 6% of the adult population in Western society. Additionally, diabetes-related complications exact a heavy toll on patients with poor metabolic control. A total of 200–300 million cases of T2DM worldwide is expected by 2010 (1), and a decisive force driving this increasing incidence is the ongoing obesity epidemic (2). Although no cure is yet available for T2DM, despite the first therapeutic administration of insulin eighty years ago, oral therapies for T2DM have been developed and are widely used. These therapeutics act by stimulating insulin release (e.g., sulfonylureas, meglitinides), by enhancing insulin action (e.g., thiazolidinediones), by delaying the digestion and absorption of polysaccharides (e.g., α-glucosidase inhibitors), or by lowering hepatic glucose production by unclear mechanisms (e.g., biguanides such as metformin) (3). These therapies, however, are not perfect and are characterized by insufficient efficacy, limited tolerability, or significant mechanism-based side effects; none of these therapies are effective in type 1 diabetes mellitus (T1DM) patients who are totally lacking insulin and many T2DM patients respond weakly or not at all. In addition, few of the available therapies adequately address underlying defects such as obesity or insulin resistance, and many patients who respond initially become refractory to treatment over time—this is particularly true with sulfonylureas. Therefore, novel treatment options are urgently needed that take advantage of physiological regulatory mechanisms and that result in weight loss or lack of weight gain.
Years of drug hunting have now paid off for Grimsby et al. (4) who recently identified the first specific glucokinase-activating agent, RO-28-1675, which augments both hepatic glucose metabolism and glucose-induced insulin secretion. These exciting new findings may open up major progress towards a truly effective treatment for diabetes, because not only for the first time two of the major pharmacological approaches have been combined in one drug, but also this drug’s action appears to be endogenously regulated by blood glucose levels.
Glucokinase, also termed Hexokinase D (EC 2.7.1.1), is the characteristic isoenzyme of hexokinase in pancreatic islet β-cells (B-type GK; GKB) and in the liver (L-Type GK; GKL) that phosphorylates glucose at the sixth carbon position. In β-cells and hepatocytes, the rate of glucose metabolism is determined mainly by the activity of GK: inactivating and activating mutations in the GK gene have been linked to maturity-onset diabetes of the young (MODY), Type 2 (MODY2), and persistent hyperinsulinemic hypoglycemia of infancy (PHHI), respectively (5 –8) (Box 1). Because of the high capacity of the facilitative glucose transporter GLUT2 (9) present in β-cells and hepatocytes, extracellular glucose concentrations are sensed intracellularly by GK (10), which determines the threshold for insulin secretion, whereas in the liver, GK facilitates hepatic glucose uptake during hyperglycemia (11) (Figure 1). A peculiar property of the monomeric GK is its cooperative kinetics with its substrate glucose [Hill coefficient of 1.7 where values of > 1 indicate positive cooperativity—that is, the binding of one ligand facilitates binding of subsequent ligands at other sites on a multimeric receptor complex; the Hill coefficient was originally worked out for the binding of oxygen to hemoglobin (Hill coefficient of 2.8)]. Most importantly, single-point mutations (V62M, D158A, Y214A, V455M, F456V) in a region distinct from the substrate binding sites of the enzyme lead to allosteric activation of GK—this unexpected finding has fueled the hunt for GK activators. Indeed, Grimsby et al. show that RO-28-1675 acts as an allosteric activator of GK (4). At micromolar concentrations in vitro, the RO-compound augments the activity of GK by increasing Vmax (~1.5 fold) and by decreasing the apparent Km for glucose (from ~8 to 2 mM) without changing the Hill coefficient or the Km for ATP. Consistent with the strong correlation between GK activity and insulin secretion (12), RO-28-1675 decreases the threshold concentration of glucose necessary for glucose-induced insulin release (GSIR) in isolated perifused rat pancreatic islets and increases the amount of maximally secreted insulin by activating glucose metabolism in the β-cells.
Disorders of glucose homeostasis.
Type I diabetes mellitus (T1DM; Insulin-dependent, IDDM) is a disorder of glucose homeostasis that results from the failure of pancreatic β-cells to synthesize and secrete insulin in response to elevated glucose levels. Type II diabetes mellitus (T2DM; Non-insulin-dependent, NIDDM) is a heterogeneous disorder of glucose homeostasis characterized by failure of both, insulin target tissues to respond to insulin and β-cells to secrete sufficient insulin. Persons with T2DM usually (Western cultures), but not always (Eastern cultures) have an obese body habitus, and manifestations of the so-called metabolic syndrome characterized by diabetes, insulin resistance, hypertension, and hypertriglyceridemia. Maturity-onset diabetes of the young (MODY) is a distinct type of NIDDM with a monogenic background and onset at age less than twenty-five years. MODY2 patients often are non-obese and lack the metabolic syndrome. MODY2 is caused by inactivating mutations in the GK gene. Persistent hyperinsulinemic hypoglycemia of infancy (PHHI) is a rare disorder arising from defective negative feedback regulation of insulin secretion by low glucose levels and has been associated with activating mutations of the GK gene, as well as mutations in the SUR–KIR6.2 potassium channels responsible for glucoseregulated insulin release.
Grimsby et al. demonstrated that in wild-type C57BL/6J mice and in several rodent models of T2DM, single-dose oral administration of RO-28-1675 reduces blood glucose levels and increases plasma levels of insulin. Orally administered RO-28-1675 also reduces postprandial (i.e., after food intake) blood glucose levels without further increasing plasma insulin levels. In fact, RO-28-1675 simultaneously targets GK in the liver, resulting in a shift from hepatic glucose production to hepatic glucose uptake and glycogen synthesis.
In the liver, GK activity is acutely regulated by glucokinase regulatory protein (GKRP), which binds and inhibits GK. Moreover, binding of GKRP to GK leads to nuclear sequestration of the GKRP–GK protein complex (13). The inhibitory effect of GKRP on GK depends on the presence of fructose-6-phosphate that competes with glucose (for binding to GK) and is antagonized by fructose-1-phosphate (14). Interestingly, RO-28-1675 reverses the inhibitory action of GKRP in vitro, indicating that the compound may also affect the subcellular localization of GK in hepatocytes.
Several GK-activating mutations (e.g., V455M and A456V) have been identified in humans who present with hyperinsulinism and hypoglycemia (7, 8). RO-28-1675 appears to bind to the very same region where these mutations occur and might mimic the effects of these amino acid exchanges. Therefore, it is tempting to speculate that a natural endogenous counterpart of the RO-compound may exist that is involved in the allosteric regulation of GK.
Until now GK was thought to be mainly a “glucose sensing” metabolic player at the level of pancreas and liver, and also the main glucose sensor in the brain (15, 16). Glucokinase, however, not only improves hepatic glycogen storage and enhances glucose-stimulated insulin release, but also appears to link glucose regulation with apoptosis. A group of scientists at Beth Israel Deaconess Center and the Dana-Faber Cancer Institute at Harvard Medical School in Boston demonstrated very recently that a pro-apoptotic BCL-2 family member named BAD (the Bcl-2/Bcl-XL– associated death promoter), triggers cell death in response to abnormalities in glucose metabolism and builds complexes with glucokinase (17). Although GK is the only hexokinase exclusively expressed in liver and pancreas, it remains unclear whether BAD can also interfere with the activity of other hexokinases. In any case, GK seems to be a far more important metabolic target than previously assumed. On the basis of the recent advances described here, perhaps even more physiological functions of and pharmacological perspectives on GK will be understood and developed. Central administration of a GK activator such as RO-28-1675 could, for example, suppress neuronal circuits in the hypothalamus and the brainstem, thereby regulating appetite and energy expenditure as a consequence of constantly activated glucose sensors in the brain despite the lack of caloric intake. In case these compounds can be transported across the blood brain barrier and bypass the central appetite regulatory systems, weight loss might be achieved and the future development of drugs to fight obesity as well as diabetes type 2 might be possible.
Nevertheless, all novel drug candidates have to pass numerous steps before becoming available for the patient, one of the most difficult tasks being the tests for potential unwanted side-effects. For RO-28-1675 and comparable GK activators, uncontrollable hypoglycemic events may turn out to become a major issue, as well as possible hepatic fat accumulation with long-term treatment based on the proposed mechanism of these agents.
From the data presented, it becomes quite clear that the β-cell appears to be the main site of action, as mice lacking insulin no longer respond to the drug. This is not surprising considering the finding that liver-specific knockout of GK in mice has only minor effects on plasma glucose levels (18). Therefore, it appears unlikely that RO-28-1675 might be beneficial for patients with T1DM or with progessive forms of T2DM where the β-cells already have failed to produce enough insulin.
Given the multifactorial nature of the genetic and environmental factors that contribute to the genesis of metabolic syndrome and T2DM, the recent research developments described above must be regarded as some of the most promising discoveries toward a future of truly effective treatment for diabetes. Further efforts to characterize disease “subphenotypes” and specific genetic markers will translate into more selective, tailor-made therapies, which are even more favorable if they can attack “two birds with one stone.”
Samuel W. Cushman, PhD, is Chief of the Experimental Diabetes, Metabolism, and Nutrition Section, Diabetes Branch, National Institute of Diabetes and Digestive and Kidney Diseases, NIH. Please address correspondence to SWC. Email sam_cushman{at}nih.gov; fax 301-402-0432. Matthias H. Tschöp, MD, is an Associate Professor at the University of Cincinnati (starting Autumn 2003) and is currently Senior Scientist at the German Institute of Human Nutrition. Hadi Al-Hasani, PhD, is an Assistant Professor at the University of Cologne and is a Senior Scientist at German Institute of Human Nutrition.
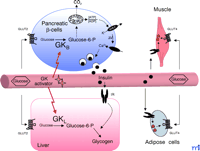
Central role of glucokinase (GK) in whole-body glucose homeostasis. In pancreatic β-cells, B-type GK (GKB) constitutes part of the glucose sensor. Glucose is transported into the cells by the glucose transporter isoform GLUT2, to be phosphorylated by GK yielding glucose-6-phosphate. Glycolysis and oxidative metabolism of glucose increases the ATP:ADP ratio, leading to inactivation of the Kir6.2 potassium channel and to subsequent depolarization of the membrane. Following influx of Ca2+ through a voltage-gated Ca2+ channel, insulin-containing storage vesicles fuse with the plasma membrane, causing the release of the hormone into the blood stream. In the liver, production of glucose-6-phosphate by L-type GK (GKL) precedes storage of glucose as glycogen, which is stimulated by insulin. In adipose and muscle cells, insulin stimulates glucose uptake and metabolism by triggering the translocation of the glucose transporter isoform GLUT4 from intracellular storage vesicles to the plasma membrane. The novel GK activators augment both glucose-induced insulin secretion in β-cells and hepatic glucose metabolism, and as a result lead to improved clearance of glucose from the blood stream.
- © American Society for Pharmacology and Experimental Theraputics 2010

