Neuronal Nicotinic Receptor Subtypes: DEFINING THERAPEUTIC TARGETS
Abstract
Humans have appreciated the beneficial properties of the tobacco plant for thousands of years. These effects include alertness, reduced anxiety, muscle relaxation, and analgesia. Yet it has been less than two decades since the central actions of nicotine have been examined in earnest for potential therapeutic applications. In fact, the cholinergic systems, in comparison to other neurotransmitter systems of the body, have been relatively poorly exploited in terms of therapeutic agents, and the muscarinic cholinergic systems have been relegated mainly to the treatment of gastrointestinal disorders and glaucoma; for the nicotinic system, antagonists are used to induce muscle paralysis during certain surgical procedures. For both families of cholinergic receptors, widespread exploitation in terms of therapeutics has been limited by significant side effect profiles associated with available cholinergic drugs.
Introduction
The history of pharmacology is largely the history of the rational study of natural plant products in biomedicine. Natural products such as atropine, muscarine, nicotine, curare, and physostigmine (eserine) were used in early pharmacological studies of the autonomic nervous system to identify the role of acetylcholine in autonomic ganglionic, postganglionic parasympathetic, and somatic neuromuscular neurotransmission. [Nicotine itself has no therapeutic indication other than for its use in the treatment of tobacco addiction (1) and perhaps for the treatment of ulcerative colitis (2).] These early studies indicated that cholinergic receptors are divided into three major subtypes: muscarinic, nicotinic ganglionic, and nicotinic neuromuscular receptors. As the concept of chemical neurotransmission was applied to the central nervous system, it was not long before the rather ubiquitous nature of cholinergic transmission in the brain was appreciated. By the 1970s, it was even clear that cholinergic neurons were important in the brain’s control of virtually every autonomic and sensory function that could be studied. Most subsequent work applied to brain cholinergic pharmacology was directed at neurons and pathways expressing muscarinic receptors. In contrast, the recurrent inhibition induced by antidromic firing of motor neuron collaterals onto intraspinal Renshaw cells represented for many years the only example of a functioning nicotinic cholinergic synapse within the CNS (3). Ligand binding methods to estimate nicotinic receptors were aided with the use of tissues derived from the electric organs from the marine species of Electorphorus and Torpedo. Up to 40% of the electroplax tissue membrane contains nicotinic acetylcholine receptors. Similar radioligand binding techniques applied to brain tissues of rodent species revealed that high densities of nicotine binding sites were localized to the thalamic habenular nucleus. Like Renshaw cell collaterals, the cells within the habenular nucleus contain the presynaptic machinery of functioning cholinergic neurons. Intracellular recordings from cells within this region have confirmed the presence of rapid postsynaptic nicotinic excitatory responses and the presence of functional nicotinic cholinergic synapses (4).
About this time, other brain regions were shown to contain nicotine-responsive cells; however, only a very few studies in the late 1980s were suggestive of potential therapeutic approaches using nicotine other than for treating tobacco addiction. With the discovery of widespread cholinergic neuronal degeneration in autopsied brain samples from individuals with Alzheimer’s disease, including decreases in the expression of both muscarinic and nicotinic receptors, it was suggested that any therapeutic approach to treating the cognitive decline would need to address both families of cholinergic receptors (5). Although sporadic preclinical studies of the analgesic or anxiolytic properties of nicotine continued (6, 7), the impetus for using nicotine therapeutically other than for smoking cessation was limited by concerns about potentially serious side effects (8). Indeed, the two factors that may have retarded advances in the nicotine field were (1) the linkage of nicotine to cigarettes and the negative publicity associated with smoking (9); and (2) the finding that doses of nicotine required to enhance memory in rats were inordinately high. The doses were reported to improve the performance of rodents (inhibitory avoidance task) ranged from 0.05–1 mg/kg (10, 11). Human doses of about 0.3– 0.6 mg/kg nicotine approximate amounts consumed by habitual cigarette smokers over an entire day (12). Administration of nicotine to rats by chronic infusion requires doses close to 5 mg/kg/ day to enhance accuracy in spatial memory tasks (13). However, studies in smokers and human volunteers indicated that much lower doses of nicotine (e.g., equivalent to the amount derived from smoking a single cigarette) could facilitate mnemonic information storage and promote state-dependent learning (14).
The results of these human studies prompted us to examine the effect of nicotine at similarly low doses (1–10 μg/kg) given to adult rhesus monkeys well trained in the performance of a computer- assisted delayed matching-to-sample task (15). These data and other subsequent clinical studies with nicotine confirmed the high potency of the drug as a cognition-enhancing agent in human and non-human primates (16–19). Indeed, low-dose chronic administration of nicotine via transdermal patch application improves cognition in individuals with Alzheimer’s disease and in individuals with the precursor syndrome characterized by mild cognitive impairment (20). Although exclusionary criteria included subjects with cardiovascular disease, side effects were not severe and the drug was generally well tolerated.
Neurochemical Road to Safer Nicotine
At about the same time that nicotine was demonstrated to improve aspects of cognition in laboratory animals, the identification of multiple receptor subtypes of brain nicotinic receptors spurred interest by several academic and pharmaceutical laboratories in the possibility that new nicotinic drugs could be developed. Prior to 1990, however, the primary evidence that different subtypes of nicotinic receptors existed in the CNS was derived from ligand binding studies. Tritium-labeled acetylcholine (in the presence of muscarinic receptor blockade) or labeled nicotine was shown to interact with saturable, high-affinity (Kd = 1–10 nM) binding sites (21). These high-affinity nicotine binding sites were shown to be anatomically distinct from binding sites that were readily labeled with α-bungarotoxin (Kd ~ 0.5 nM), a peptide derived from snake venom that produces effective blockade of skeletal neuromuscular nicotinic receptors. Sites labeled by [125I]- α-bungarotoxin initially were considered to be “non-neuronal” nicotinic receptors because they were not always within the synapse and because the toxin failed to block the effects of nicotine in several functional assays (22).
The first breakthrough at the biochemical level in the rational targeting of central nicotinic receptors came after the structure of the muscle nicotinic receptor was elucidated (23). The receptor was shown to comprise a pentomeric transmembrane complex of α1, β1, γ, δ, or εsubunits. Each subunit consists of a four-transmembrane spanning protein with both N- and C-terminal extracellular domains. In brain, only α and β subunits exist, although there are eight unique α and three β subunit isoforms. Subunits α2, α3, α4, α5 and α6 (α2–6); and subunits β2–4 can form hetero- oligomeric nicotinic receptors, whereas α7–9 subunits can form homo-oligomeric nicotinic receptors. The α subunit contains the ligand binding domain. By expressing multiple combinations of the subunits in cultured non-neural cells, the functional combinations were identified by measuring channel properties after exposure to ligand (24).
The most efficacious of these artificially formed receptors have been compared functionally to the most prevalent nicotinic receptor subtypes found in the brain and autonomic ganglia. Ganglionic nicotinic receptors contain α3 and α5 subunits, which can associate with β2 or β4 subunits. In the brain, there is a differential expression and distribution of nicotinic receptors, although the exact subunit stoichiometry for each nicotinic receptor subtype is not always known. From a drug discovery viewpoint, three brain nicotinic receptor subtypes have been addressed: (1) the α4β2 subtype, which accounts for most of the high affinity [3H]-nicotine binding in the CNS; (2) the α3β4 subtype, which can be identified by [3H]-epibatidine binding (provided that the α4β2 sites are first masked with the ligand cytisine); and (3) α7 homooligomeric nicotinic receptors, which account for most of the high-affinity α-bungarotoxin binding in the CNS. Unlike the family of muscarinic cholinergic receptors, nicotinic receptors are more widely dispersed and do not always conform to classical cholinergic pathways; however, the highest expression of α4β2 nicotinic receptors occurs in the thalamus, cerebral cortex, and dorsal hippocampus. As suggested by their anatomical location, functional studies have shown that the α4β2 subtype mediates aspects of cognition and memory and participates in sensory gating (Figure 1⇓). Subunit compositions containing the α3 subunit (as the sole α subunit or in combination with α5 or α6) and the β4 (or β2) subunit are even less discretely localized than are the α4β2 nicotinic receptors; however, the α3β4 subtype is highly expressed within the medial habenula and the interpeduncular nucleus, and also within the optic tract, cerebellum, and certain cranial nerve nuclei (25). Subtype compositions containing α3 and α6 subunits were initially regarded as receptors that, from a therapeutic standpoint, should be avoided; specifically, the α3β4 subtype was thought to be highly expressed within the striatum and the nearby nucleus accumbens, where they could mediate the release of dopamine produced by nicotine. Whereas dopamine release within the striatum could have a potential therapeutic role for the treatment of Parkinson’s disease, dopamine release within the nucleus accumbens is considered to underlie the rewarding effects of nicotine. Subsequently, however, α6α3β2 rather than α3β4 receptors appear to be the dominant α3-containing receptors within the accumbens (25); α4β2 nicotinic receptors, moreover, may yet prove even more relevant for nicotine’s reinforcing actions (26). The α3β4 nicotinic receptor still may be a subtype to avoid, because it mediates autonomic ganglionic transmission, and drugs acting on the α3β4 subtype may mediate the cardiovascular side effects of nicotine.
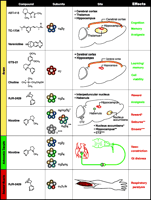
Subtypes of nicotinic acetylcholine receptors present in brain, autonomic ganglia, and the neuromuscular junction. Compounds designed to preferentially activate the given subtypes are discussed in the text. The anatomical locations are based on agonist-induced physiological responses. “Effects” given in green represent the beneficial actions of nicotinic receptor agonists, whereas “effects” in red represent undesirable actions or side effects. The colorcoding of receptor subunits does not represent actual subunit stoichiometry within given receptor type. (CTZ, chemoreceptor trigger zone.)
As mentioned above, the homo-oligomeric α7 nicotinic receptor is very unlike the heteromeric subtypes, particularly with regard to its low affinity for nicotine. The initial identification of the α7 subtype as an authentic nicotinic receptor was made through its binding to the skeletal muscle toxin α-bungarotoxin; it was perhaps the similarity of the α7 nicotinic receptor to the muscle nicotinic receptor that formerly excluded the subtype from therapeutic targeting. The brain distribution of α7 nicotinic receptors is primarily restricted to certain layers of the cerebral cortex and hippocampus, as well as to the hypothalamus (27). The α7 nicotinic receptor is thus a potential therapeutic target, largely because of its strategic localization within the hippocampus, suggesting a role in cognition and memory; the extrasynaptic localization of the α7 receptor subtype reflects its extended role in cell viability.
Road Obstacles
Elegant molecular experiments (described above), coupled with studies of anatomical and ultrastructural localization, have established a starting point for the rational design of novel analogs of nicotine. The objective thus continues to be the discovery of novel drugs that feature the beneficial actions of nicotine while eschewing its considerable side effect profile. Presently, new nicotine-like compounds may be screened in binding assays in which an individual receptor subtype is expressed in cultured non-neural cells; functional efficacy can be assessed by measuring membrane currents in transfected cell lines. However promising a drug candidate might seem after passing these initial screens, several idiosyncratic features of the nicotinic cholinergic system may conspire to relegate a potential therapeutic compound to the storage shelf. Already mentioned is the relatively even brain expression of nicotinic receptor subtypes, which increases the difficulty of targeting a subtype-selective drug to a specific brain structure (although brain distribution may be altered in disease states such as Alzheimer’s disease). Also, it is not yet clear which specific subtypes would, upon activation, provide optimal efficacy. For the heteromeric receptor subtypes, it has been notoriously difficult to develop high subtype selectivity. This difficulty may arise because selectivity is largely conferred by the β subunit, and there are only two subunit isoforms (i.e., β2 and β4) that appear to have physiological significance.
Another feature of nicotinic pharmacology is the ability of the receptor to rapidly desensitize, limiting the action of an agonist. It has been suggested, for example, that cigarette smoking causes a cycle of receptor activation, desensitization, and re-sensitization, over a time course consistent with the urge to resume smoking. In other situations, nicotine-induced desensitization may confer desired physiological outcomes. The desensitization of nicotinic receptors usually occurs at concentrations of nicotine much lower than those required for inducing an agonist-like response (e.g., neurotransmitter release); these lower desensitizing concentrations of nicotine are more commensurate with the in vivo doses required for cognition enhancement. One explanation for this beneficial desensitization may be the concomitant loss of tonic cholinergic modulation of secondary (possibly GABAergic) inhibitory interneurons. Finally, nicotine and certain of its analogs appear capable of inducing a protracted degree of improvement in tasks that involve cognition. Because cognition enhancement is a primary indication for nicotinic drugs, the persistence of drug efficacy beyond the drug’s presence in blood and brain is problematic for determining an appropriate dosing schedule.
Rational Approach to Nicotine Drug Design
A rational approach to the design of novel nicotinic receptor agonists began in full swing during the early 1990s. Many compounds were shown to exhibit more favorable side effect and toxicity profiles relative to nicotine in animal models. Many were also reported to exhibit therapeutic potential in preclinical studies. ABT-418, (S)-3-methyl-5-(1-methyl-2-pyrrolidinyl) isoxazole, was one of the first in a series developed by Abbott Labs. The compound exhibited some favorable subtype selectivity for the α4β2 receptor over the α7 receptor. Our studies of the drug in monkeys confirmed the enhancement of cognitive performance with potency and effectiveness similar to nicotine. Indeed, ABT-418 significantly improved accuracy by macaques in the performance of a delayed matching-to-sample task. The improvement in task performance was evident in both young and aged monkeys, and following both intramuscular and dermal patch administration; there was no evidence of tolerance (28, 29). Significantly, the drug was able to reverse the decrease in accuracy caused by a task-relevant distractor used to provide a primate model for attention deficit hyperactivity disorder (ADHD) (30). ABT-418 also has been evaluated in the clinic, and the most recent studies continue to show effectiveness in individuals with Alzheimer’s disease, Parkinson’s disease, and ADHD (31–33). However, the drug’s short duration of action did require a dermal patch formulation, which has its own advantages and disadvantages. In contrast, Abbott’s potent nicotinic receptor agonist ABT-594 [i.e., (R)-5-(2-azetidinylmethoxy)- 2-chloropyridine)] was initially developed as an analgesic drug. Although equally effective as the high-affinity agonist epibatidine in preclinical tests for analgesic activity, ABT-594 did not exhibit the toxicity elicited by epibatidine in the therapeutic range. The greater safety profile for ABT-594 was attributed to preference for the α4β2 subtype of the nicotinic receptor (34). The compound was in phase-II clinical evaluation in 2001, and it is still being investigated.
Using proprietary nicotinic receptor–expressing cell lines, SIBIA Neurosciences entered the nicotinic drug development fray with their lead compounds SIB-1553A [i.e., (±)-4-{[2-(1-methyl- 2-pyrrolidinyl)ethyl]thio}phenol hydrochloride] and SIB-1508Y [(S)-(-)-5-ethynyl-3-(1-methyl-2-pyrrolidinyl)-pyridine]. SIB- 1553A predominantly activated subtypes possessing β4 subunits (e.g., α2β4), whereas SIB-1508Y exhibited selectivity for α4β2 receptors (35). These differences in receptor subunit selectivity manifested differing profiles of neurotransmitter releasing activity. Although both compounds, like nicotine, could induce striatal dopamine release, SIB-1553A was significantly more effective than either SIB-1508Y or nicotine in evoking hippocampal acetylcholine release. These neurotransmitter release experiments continued to support the concept that nicotinic receptor agonists with differing preferences for activating certain subtypes of nicotinic receptors could exhibit both anatomical and functional specificity. The question remained as to whether this level of specificity would be apparent in whole animal studies. In fact, SIB-1553A was earmarked primarily for the treatment of Alzheimer’s disease, and SIB-1508Y was expected to be superior in treating Parkinson’s disease; the two syndromes manifest, respectively, brain cholinergic and dopaminergic deficits. The expectations for the two drugs in this regard were borne out in non-human primates (36, 37). As with nicotine, these compounds exhibited therapeutic potential in animal models of neuropsychiatric disorders. Like ABT-418 mentioned above, SIB-1553A reversed distractor-induced accuracy in a delayed matching task by young monkeys (38). SIB-1508Y enhanced vigilance, reversing the known attention deficits in aged rats (37), and alleviated a learned helplessness task (39). In early 1999, SIB-1508Y was evaluated in two phase-II clinical trials; in one trial, the compound was combined with other anti-Parkinson’s agents, whereas monotherapy for early-stage Parkinson’s disease was employed in the second trial. In mid 1999, a phase-II study of SIB-1553A was initiated in individuals with Alzheimer’s disease. The results of these studies have not yet been published.
Taiho Pharmaceutical Company, along with a team of academic researchers from the University of Florida, Gainesville, introduced an alternate structure to the mix of new nicotinic receptor drugs. Their compound, GTS-21 [i.e., 3-(2,4-dimethoxybenzylidene) anabaseine, was patterned after the invertebrate toxin anabaseine, a nonselective nicotinic agonist. GTS-21 and its active metabolite 4-0H-GTS-21 have been characterized pharmacologically as partial agonists with preference (relative to nicotine) for the α7 subtype of the nicotinic receptor (40). Although the drug also interacts with equal or greater affinity to α4β2 nicotinic receptors, it appears to display the functional characteristics of an α7 partial agonist. Despite its completely different subtype profile from the aforementioned nicotinic receptor agonists, GTS-21 was also shown to have cognitive enhancing actions with a low potential for side effects relative to nicotine (41). A recent clinical safety trial with GTS-21 in normal healthy males reveals that the compound is well tolerated and that it enhances attention and working memory (42). What sets this compound apart somewhat from the heteromeric subtype-preferring agonists is perhaps the relative specificity of action for the drug in terms of cognition enhancement. Specifically, GTS-21 is not active in behavioral tasks in which nicotine is effective (43). Thus, the potential for GTS-21 as a therapeutic agent for Parkinson’s disease, or for attention deficit disorders, anxiety disorders, depression, or schizophrenia may be limited relative to heteromeric subtype–preferring agonists. Of particular interest is the failure of virtually all of the subtypepreferring agonists discussed thus far to substitute for nicotine in paradigms in which animals are trained to respond to the rewarding effects of nicotine. So, it seems that this one large hurdle, the addictive risks connected with cigarette use, may be overcome.
Targacept, a pharmaceutical company spun off from RJR Tobacco Company, in 2000, has developed a number of compounds relevant to the nicotinic receptor field. The company focuses on a broad spectrum of disease entities, including Alzheimer’s disease, cognitive impairment in the elderly, attention deficit disorders, pain, depression/anxiety, and schizophrenia. One of their first potentially subtype-selective compounds was the substituted pyridine derivative RJR-2429 [i.e., (6)-2-(-3-pyridinyl)- 1-azabicyclo[2.2.2]octane]. The compound exhibits preference for striatal α4α3β2 nicotinic receptors, although it manifests overlapping affinity for both the ganglionic (α3β4) and the muscle subtypes (44). RJR-2429 has been used as a pharmacological tool rather than a drug entity; however, Targacept followed up with a series of new analogs over the past few years. Table 1⇓ presents a partial listing of novel nicotinic receptor agonists, their disease targets, receptor subtype preference, and status in the development pipeline. Of particular interest is compound TC-1734 [i.e., 2S-4EN- methyl-5-(5-isopropoxy-3-pyridinyl)-4-penten-2-amine], which is currently in phase-II clinical trials for cognitive enhancement in the elderly. The compound has very good affinity for the α4β2 receptor subtype (Ki = 11nM) but poor or no interaction with α7, ganglionic, and neuromuscular nicotinic receptor subtypes (45). In rats, TC-1734 reverses the amnestic actions of scopolamine, and enhances the procognitive actions of the Alzheimer’s drug donepezil. The drug is also effective in improving the performance of adult rats in the radial arm maze, a spatial working memory task. Intiguingly, TC-1734 prolongs duration of positive mnemonic responses in this task. Task accuracy is improved for up to eighteen hours following a single administration, far exceeding its plasma half-life. The ability of certain cognition-enhancing drugs to evoke prolonged pharmacodynamic effects in experimental animals performing memory-related tasks may prove to be an important component of their pharmacology (46). The effect certainly makes decisions regarding dosing schedules more complicated.
Nicotinic compounds under development
First to Market
One of the first nicotinic receptor agonist other than nicotine to reach the U.S. market is Janssen Pharmaceutica’s Alzheimer’s drug galantamine. Galantamine (Reminyl®), a plant alkaloid formerly known as galanthamine, is a cholinesterase inhibitor that also can act as a noncompetitive agonist at nicotinic receptors (47). Nevertheless, there continues to be debate as to the extent to which nicotinic receptor activation plays a role in the drug’s profile of therapeutic benefit in slowing the progression of Alzheimer’s disease (48).
Pfizer Inc. will likely boast the first synthetic nicotinic receptor agonist to debut in the U.S. market. The company’s new antismoking therapy, varenicline [i.e., equal admixtures of 6,10-methano- 6H-pyrazino[2,3-h][3]benzazepine and 7,8,9,10-tetrahydro-, (2R,3R)-2,3-dihydroxybutanedioate], already reached phase-III clinical trials. Varenicline is a partial agonist that is specific for the α4β2 nicotinic receptor subtype; partial agonism may conceivably reduce nicotine craving without itself being rewarding or addictive. The results of early clinical trials, involving several hundred smokers, indicate that almost half of the study participants treated with oral varenicline are able to quit smoking after seven weeks. If confirmed, these data are the most robust of results yet attained in a well-controlled smoking cessation study. Varenicline’s introduction to the world market would solidify the proof-of-concept for a “safe” and non-addictive nicotine-like compound and will lend credence to the use of synthetic nicotinic receptor agonists for other indications.
Alternate Approaches to the Design of Nicotinic Receptor Agonists
Choline-Derived Agonists
Analogs of choline, the proximate precursor for the neurotransmitter acetylcholine, have been studied intermittently over the past thirty years. Several choline analogs, both N-substituted as well as N-replacement analogs (e.g., arsenonium, sulfonium, and selenonium), have been shown to be substrates for the plasma membrane high-affinity choline transporter; they are thus transported into the cell, where they are acetylated and stored in synaptic vesicles, and the acetylated analog is released upon nerve stimulation. A new rationale has emerged for revisiting these choline analogs for use as therapeutic agents. Choline itself has been demonstrated to be a full and selective agonist at the α7 nicotinic receptor (49, 50) and a co-agonist (with acetylcholine) at the α4β2 receptor (51). In fact, choline is only about tenfold less potent than acetylcholine in its ability to activate α7 nicotinic receptors. The rationale for targeting the α7 subtype derives from the effectiveness of GTS-21 as a cognition-enhancing drug, as well as from studies indicating that nicotine’s neuroprotective actions may be mediated in part via stimulation of α7 receptors. Indeed, the neuroprotective potential of nicotine is almost incontrovertible (52, 53). Choline, like nicotine, can protect neural cells from cytotoxicity induced by growth factor deprivation (54) or by exposure to the glutamate analog AMPA (53). Choline itself is of limited clinical usefulness due to its mM affinity for α7 nicotinic receptors and its reduced efficacy as cytoprotectant relative to nicotine (55). Also, in early clinical trials in which “precursor-loading” with choline was examined as a potential Alzheimer’s disease treatment, the drug was associated with poor compliance due to its malodorous bioconversion in the gastrointestinal system to trimethylamine.
We have examined a number of choline analogs using, as a model system, cultured differentiated PC- 12 (sympathetic cholinergic neuronal) cells in which cytotoxicity had been induced by the withdrawal of trophic factors (nerve growth factor and serum). Analogs of choline were synthesized initially based on the greater cytoprotective potency and agonist efficacy relative to pyrrolidinecholine (56); the cytoprotective activity of pyrrolidinecholine is known to be mediated by the α7 subtype of nicotinic cholinergic receptors. Yet as a cytoprotective agent, and as an agonist for the α7 subtype of nicotinic receptor, pyrrolidinecholine is less potent than nicotine; furthermore, the analog would require active transport via a choline carrier into the brain. We have developed choline analogs with similar or greater potency than nicotine. The chemical structures of six novel analogs (and one commercially available analog) are depicted in Figure 2⇓. All are tertiary amines, a structural requirement that allows for penetration of the drug into the CNS without the need to be transported by the blood to brain choline transporter. All of the compounds produce some level of cytoprotection, although there are clear differences in potency and efficacy. The “concentration – effect” curves for the analogs are very similar to nicotine’s biphasic profile. JAY-2-21-29 and JWB 1-85-1 manifest potency and efficacy comparable to nicotine. In fact, JAY-2-21-29 is most potent, with an ED50 of approximately 30 nM. Pretreatment with the α7 subtype–selective nicotinic receptor antagonist methylylcaconatine, furthermore, completely prevents the cytoprotective action produced by JAY 2-21-29. Pretreatment with the β2 subtype- preferring antagonist DHβE was without effect in this regard.
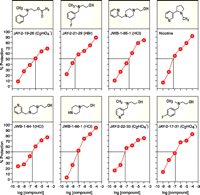
Cytoprotection afforded by choline analogs. Data represent the ability of nicotine and 7 tertiary amine analogs of choline to protect differentiated PC-12 cells from the cytotoxicity associated with growth factor withdrawal. Horizontal and vertical lines within each graph show concentrations that afford 50% cytoprotection.
Analogs of choline thus have the potential to interact with α7 nicotinic receptors. Additional components to the therapeutic picture may be derived from those analogs that may also serve to enhance cholinergic synaptic efficiency as false neurotransmitters. Synaptic efficiency might be enhanced if the choline analog is acetylated in vivo and released as a false transmitter with enhanced affinity for the postsynaptic cholinergic receptor, and/or resistance to acetylcholinesterase (Figure 3⇓). Further experimentation will be necessary to determine whether acetylation of choline analogs would result in higher affinity for postsynaptic cholinergic receptors or in compounds resistant to turnover by acetylcholinesterase.
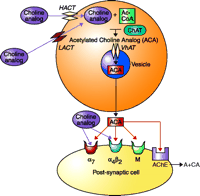
Representation of the cholinergic synapse. The potential sites of action for novel analogs of choline are indicated. The choline analog may be transported into the cell by the high-affinity (HACT) or low-affinity (LACT) choline transporters, so that the drug could compete with endogenous choline for acetylation by acetylcoenzyme-A (Ac-CoA) and the synthetic enzyme choline acetyltransferase (ChAT). The acetylated choline analog (ACA) would then be transported into the cholinergic synaptic vesicle by the vesicular acetylcholine transporter (VhAT). Cell depolarization resulting from neural activity would cause the vesicular release of the ACA (i.e., a “false neurotransmitter”). Within the synapse, the ACA could interact with nicotinic or muscarinic (M) receptors. The ACA also could interact with soluble or membrane-bound acetylcholinesterase (AChE), the enzyme that normally degrades ACh. If the ACA either inhibits AChE or is a poor substrate for hydrolysis by AChE, synaptic efficiency would be increased. Alternatively, the choline analog could directly activate nicotinic receptors, particularly the α7 subtype.
Cotinine
The development of novel synthetic nicotinic receptor agonists will be informed by the structural requirements for receptor subtypes as gleaned from natural products including cytisine, anabaseine, lobeline, galantamine, and choline. With the possible exception of choline, none of these compounds exhibit a toxicity profile significantly better than that for nicotine. Cotinine, the principal metabolite of nicotine, enjoys a prolonged plasma half-life (fifteen to nineteen hours) with respect to nicotine. Evidence is sparse, but there are indications that cotinine is a pharmacologically active substance associated with tobacco use. Based on multiple studies in animals and in humans, cotinine likely exerts measurable effects on behavior and cognitive function. Cotinine does not induce nicotine-like craving behavior, and the compound is relatively safe to administer at doses up to ten times that usually obtained during cigarette smoking (57).
In binding studies, nicotine is about 100-fold more potent than cotinine in displacing [3H]-epibatidine from rat cortical membranes (58). In functional assays (59, 60), cotinine exhibits the properties of a weak α7 nicotinic receptor agonist. Preliminary studies from our laboratory have confirmed that cotinine possesses at least three nicotine-like properties (61): (1) In the rat, the motor response to acoustic startle can be inhibited by the presentation of a low-level acoustic prepulse presented just in advance of the high-level acoustic pulse; the ability of the CNS to modulate its sensitivity to a sensory stimulus in this way is known as “sensory gating.” Drugs like apomorphine, which can induce a schizophreniform action can disrupt sensory gating, and under the conditions established at baseline, apomorphine treatment thus suppresses prepulse inhibition of the acoustic startle response. Most drugs with potential antipsychotic actions reverse the effects of apomorphine. Treatment with cotinine significantly reverses the effects of apomorphine on acoustic startle, supporting the possibility that the metabolite may share antipsychotic properties with nicotine. (2) Cotinine is effective in preventing the cytotoxicity associated with growth factor withdrawal in differentiated PC-12 cells. In this regard, cotinine is slightly more potent than nicotine. (3) Cotinine produces a dose-dependent increase in accuracy in an automated delayed matching-to-sample task in macaques; cotinine is only about 30-fold less potent than nicotine. This preliminary study has been extended, and data are presented in Figure 4⇓. Overall, the data provide evidence for the ability of cotinine to evoke a nicotine-like pharmacological response in three distinct model systems. Of particular surprise was the marked potency of the drug relative to nicotine in the cytoprotection assay. Although this in vitro model does not address the effects of nicotine metabolism into cotinine, the data support the possibility that the neuroprotective actions attributed to nicotine in vivo might be at least partly due to the presence of its metabolite in the CNS for prolonged periods after nicotine consumption. Data further suggest (Figure 4⇓), but do not prove, the contention that nicotine’s major metabolite, cotinine, mediates the protracted mnemonic effects of nicotine. Some consideration might be given to the development of cotinine-like compounds for use as neuroprotective agents. The drug has been shown to be safe in rather high doses in humans, and it does not appear to possess the rewarding properties of nicotine. Moreover, cotinine’s long duration of action makes the drug a good candidate as a therapeutic agent. It is intriguing that cotinine, which represents only a very small difference in chemical structure from nicotine, should exhibit such comparatively favorable therapeutic potential.
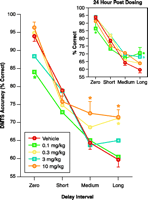
Cotinine-mediated task performance in Rhesus monkeys. Delayed matching-to-sample (DMTS) task performance was initiated ten minutes following the intramuscular administration of cotinine. The inset shows results of DMTS sessions run twenty-four hours after cotinine administration. Asterisks indicate significant differences (P<0.05) from mean accuracy values obtained after administration of drug vehicle (sterile normal saline). The results derived from seven Rhesus monkeys support preliminary studies indicating that cotinine-enhanced task accuracy in a dose-dependent manner, particularly during Long-delay trials (most difficult). Unexpectedly, the lowest dose (0.1 mg/kg) significantly decreased accuracy during Zero-delay trials (near simultaneous matching). Usually, a decrease in the performance of the shortest-delay trials suggests a decrease in attention. The particular improvements obtained during Long-delay trials suggest that cotinine, like nicotine, can augment aspects of retrieval or recall. Also, like nicotine, the positive mnemonic effects of cotinine extended to the day after drug administration (see inset). Even the decrease in accuracy during Zero-delay trials produced by the 0.1 mg/kg dose was evident.
Conclusions
A very large proportion of the worldwide drug discovery effort is being directed at the elucidation of novel G protein–linked receptors. The anticipation is that the human genome will be “mined” for new members of this ubiquitous class of cell-signaling molecules and the potential disease targets they might represent. Because G protein–linked receptors currently serve as targets for a large proportion of currently prescribed “blockbuster” drugs, this research effort appears to be well justified. The exploitation of ligand-gated ion channel receptors is perhaps not as appealing, because full agonists can alter the main flow of chemical information throughout the nervous system, so that their pharmacological actions occur with very small margins of safety. However, the discovery that multiple combinations of individual subunits make up the main classes of ligand-gated ion channel receptors including glutamate, GABA, glycine, and nicotinic receptors now permits the targeting of subtypes of these receptors, thereby allowing for anatomical and functional diversity. The examples of new nicotinic cholinergic receptor agonists cited above support this concept quite well. Novel synthetic nicotinic receptor agonists have been demonstrated to exhibit preferable, and in many cases acceptable, side effect profiles relative to nicotine. Even those compounds that exhibit little subtype selectivity in binding assays can oftentimes provide functional selectivity in whole-organ or animal experiments. The explanation for this apparent discrepancy may reside in the nature of the agonist-receptor interactions; an agonist at one subtype may show partial agonism, or even antagonism at other subtypes. In addition, the rate of desensitization and inactivation induced by a receptor agonist may dictate functional outcome.
Still, our relatively poor understanding of the nature of the pharmacodynamic effects induced by nicotine and certain related compounds that outlast the drugs’ presence in the body remains an important challenge. Until the biology of brain nicotinic receptors is more fully elucidated, the prediction of pharmacological actions, by modeling in vitro or in silico, may prove difficult. The use of animal models in a systems approach to drug discovery will continue to be necessary for the foreseeable future. But with the enormous therapeutic potential for the class of drugs acting on nicotinic receptors, our ongoing efforts to study this group of important receptors, from a wide variety of experimental perspectives, are well justified.
Acknowledgments
The studies described as from the author’s laboratory were supported by the Alzheimer’s Association, the Institute for the Study of Aging, the Veterans Administration, the Philip Morris External Research Program, Abbott Labs, and SIBIA Neurosciences, Inc.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Jerry Buccafusco, PhD, is Professor of Pharmacology and Toxicology and Professor of Psychiatry and Health Behavior at the Medical College of Georgia, and Director of the College’s Alzheimer’s Research Center. He holds a co-appointment as Research Pharmacologist at the Augusta Veterans Administration Medical Center, and is Past- (and founding)-Chair of the ASPET Division for Systems and Integrated Pharmacology. E-mail jbuccafu{at}mcg.edu; fax 706-721-9861.

