REGULATORs OF G PROTEIN SIGNALING & DRUGS OF ABUSE
Abstract
Drugs of abuse such as opioids and stimulants share a common dopaminergic reward pathway; however, in response to continual intermittent exposure to such drugs, there are neuronal alterations leading to changes in behavior. Regulators of G protein signaling (RGS) are proteins that negatively regulate G protein signaling and are expressed in brain areas important for the pharmacology of abused drugs. Moreover, the level of expression of several of these proteins is regulated by abused drugs. In this article, we discuss RGS proteins, their regulation by morphine and stimulants, and how altered levels of these proteins affect cell signaling to contribute to the pharmacology and behavioral consequence of abused drugs. Finally, we consider if RGS proteins represent viable targets for drug abuse medications.

Introduction
Drugs and other abused substances have many different primary targets, including opioid receptors, nicotinic receptors, cannabinoid receptors, monoamine transporters, and benzodiazepine receptors. Many of these drugs share a common effect, namely, to increase dopaminergic signaling in regions of the mesolimbic dopaminergic system (1), which projects from the ventral tegmental area (VTA) to the nucleus accumbens, caudate putamen, olfactory tubercle, frontal cortex, and amygdala. This increased dopamine signaling underlies the rewarding effects of the abused drugs and contributes to drug-seeking behavior (2). Stimulants such as cocaine and amphetamine increase dopamine levels by enhancing the release and/or blocking the re-uptake of dopamine through the dopamine transporter. Most other abused drugs indirectly increase dopamine release. For example, opioid drugs that act on μ-opioid receptors, including heroin, disinhibit GABA neurons that would otherwise inhibit dopaminergic neurons in the VTA; this disinhibition increases release of dopamine in the nucleus accumbens and other regions of the limbic forebrain (3, 4). Nicotine and alcohol also increase dopamine in the nucleus accumbens, possibly by activating endogenous opioid pathways (2). Correspondingly, dopamine receptor antagonists attenuate the behavioral and reinforcing effects of cocaine (5) and morphine (6), and the rewarding effect of cocaine is eliminated in dopamine and serotonin transporter knockout mice (7). There is also crosstalk among the systems that are central to drug abuse. In rats, μ-opioid receptors in the nucleus accumbens, caudate putamen, and amygdala are increased by a pattern of bingeing on cocaine (8), an effect also seen in human cocaine addicts (9).
With habitual, intermittent exposure to drugs of abuse, adaptive changes in sensitive neuronal systems lead to tolerance, sensitization, and dependence. Tolerance is a loss of responsiveness that requires escalating doses in order for the organism to achieve a given pharmacological effect; sensitization is an increase in responsiveness following multiple exposures and is of prime importance in drug craving for stimulants; and dependence is defined by withdrawal symptoms when drug taking ceases. These long-term changes, reflecting brain plasticity, arise because repeated exposure to drug repetitively activates signaling pathways, altering rates of gene transcription and neuronal activity. Drugs of abuse rapidly induce several members of the fos family of immediate early–onset genes in the nucleus accumbens and caudate putamen (10), for example, and the cAMP response element–binding protein (CREB) plays a role in drug-mediated gene regulation (11–13). Adaptive changes occur not only in regions directly involved in the reward pathway, but in many brain structures, including those that mediate learned or conditional responses such as the amygdala, hippocampus and cerebral cortex; indeed, changes induced by abused drugs have been likened to memory changes. Drug-induced changes in the locus ceruleus, a principle source of noradrenergic innervation, are associated with opioid physical dependence and the manifestation of symptoms of withdrawal (11).
Regulators of G protein signaling (RGS) are an important family of proteins that negatively modulate signaling through G protein–coupled receptors (GPCRs). Several members of this family are highly expressed in mesolimbic brain regions associated with reward; moreover, this expression changes in response to exposure to opioids, cocaine, and amphetamine, sometimes very rapidly. In this article, we review evidence for the involvement of three RGS proteins (i.e., RGS2, RGS4, and RGS9-2) in the actions of abused drugs.
RGS Proteins: Expression in Drug-Sensitive Neuronal Systems
Most drugs of abuse activate GPCRs, doing so either directly (e.g., opioids and cannabinoids) or indirectly (e.g., amphetamine, cocaine, and nicotine). A recent advance in the understanding of GPCR signaling was the discovery of the inhibitory role of RGS proteins (14–19). The primary mechanism of RGS protein function was determined nearly a decade ago: RGS proteins dramatically accelerate the rate of GTP hydrolysis catalyzed by heterotrimeric G proteins (20–24) (Figure 1⇓). [RGS proteins can thus be categorized as GTPase-accelerating proteins (GAPs).] Because the activation of G proteins is a balance between the rates of receptor-stimulated binding of GTP on one hand, and rates of hydrolysis of GTP on the other, RGS proteins are intimately involved in determining the efficiency of GPCR signaling (25), limiting the steady-state signal strength (26–29).
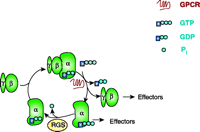
The G protein cycle. RGS proteins are GTPase-accelerating proteins (GAPs), accelerating the hydrolysis of GTP bound to Gα. RGS proteins thus reduce the lifetime of active Gα–GTP and βγ dimers, so that signaling pathways downstream of the G protein are held in check.
In humans, there are at least twenty well-characterized RGS proteins (Table 1⇓), the majority of which are “classical” in that they act on nearly all of the Gα subunits in the αi and/or αq family. Nevertheless, the R7 family of RGS proteins are selective for Gαo, although they interact to some degree with αi subunits (30, 31). This selectivity may be of key importance with respect to drugs of abuse, because D2 dopamine receptor function has been attributed largely to actions on Gαo (32). Also, RGS2 is quite selective for Gαq, although in some studies it has been shown to have some activity at Gαi (33, 34).
Mammalian RGS Proteins
Besides the classical RGS proteins, two other families of proteins with RGS homology (RH) clearly modulate Gα functions. The RH domain–containing Rho guanine nucleotide exchange factors (RhoGEFs) bind selectively to Gα12 and Gα13 and also appear to interact with Gαq (35–38). Although the RH-RhoGEFs can promote the GTPase activities of Gα12 and Gα13, the primary function of the Rho–Gα interaction is not to inhibit Gα-mediated signaling, but rather to direct GPCR-initiated informational flux into Rho-mediated pathways (39–43). Another RH domain–containing protein family, which clearly functions to inhibit Gα, comprises the GPCR kinases (GRKs). The RH domain in GRK2 binds to Gαq and inhibits its ability to activate phospholipase C, again likely not primarily by effecting a GAP function, but rather by interfering with PLC–Gαq binding. The recently solved crystal structure of GRK2 suggests a trifunctional inhibitory mechanism for the protein: first, its kinase activity inhibits GPCR signal transduction; second, its RH domain binds to Gαq; and finally, it interacts through a PH+ domain with the Gβγ dimer, blocking G protein subunits from their downstream targets (44, 45).
In addition to their Gα subunit specificity, RGS proteins may manifest receptor specificity. Wilkie and Muallem have reported that RGS4 functions preferentially at muscarinic, as opposed to cholecystokinin, receptors (46). Evidence also suggests that RGS3 shows specificity for m3 muscarinic receptors, whereas RGS5 preferentially affects angiotensin AT1a receptor signaling (47). RGS2 and RGS4 under certain circumstances may bind more tightly to the m1 and m5 receptors than to the m3 receptor (48), and RGS8 may be more active at m1 and m5 receptors than m3 receptors (49). (We will return below to the intriguing possibility of RGS selectivity for receptor subtypes, which clearly deserves additional study.)
RGS proteins may also be regulated by phosphorylation (50–52) and lipid modification (53), and they can be targeted to cellular membranes by interacting with other proteins (54, 55). A novel mechanism of regulation has been defined for RGS4 and other proteins in the R4 family. Strongly acidic lipids, such as phosphatidyl inositol triphosphate (PIP3) and phosphatidic acid, inhibit the ability of RGS4 to activate Gα GTPase activity, and this inhibition can be reversed by calcium/calmodulin (56, 57). This mechanism appears to underlie the calcium-dependent kinetics of GPCR-coupled inwardly rectifying K+ channels in the heart (58).
Whereas RGS proteins show some selectivity for GPCRs, a major determinant of their selective functionality may well be their distribution. RGS protein mRNA is generally expressed widely throughout the brain, but the distribution is not uniform, and mRNA for several RGS proteins is highly expressed in regions implicated in the action of abused drugs (Figure 2⇓). RGS2 mRNA is found in the periform and neocortex, caudate putamen, amygdala, hippocampus, and locus ceruleus (59, 60). RGS4 mRNA is found in highest levels in the cortex, thalamus, caudate putamen, and nucleus accumbens, and frequently colocalizes with dopamine receptor D2 mRNA (61, 62). RGS9 exists in two forms; the short form, RGS9-1, is only found in the retina, whereas the longer variant, RGS9-2, is almost exclusively found in striatal regions of rat and mouse brain (i.e., the caudate putamen, nucleus accumbens, islands of Calleja, and olfactory tubercle) (61, 63–65). This distribution is conserved in human brain (63) and is similar to that of other striatal specific proteins, including dopamine D1 receptors, adenylyl cyclase type V, and the phosphoprotein DARPP-32 (i.e., dopamine and cyclic adenosine 3′,5′-monophosphate regulated phosphoprotein 32kDa) (66), which suggests a role for RGS9-2 in modulating dopaminergic transmission involved in the rewarding effects of drugs of abuse. Not surprisingly, this suggestion has incited speculation that RGS9-2 could become a target in the treatment of drug abuse (18, 67).
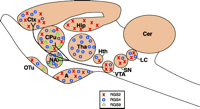
The mesolimbic dopaminergic pathway and approximate distributions of RGS protein RGS2, RGS4, and RGS9-2. (A, amygdala; CPu, caudate putamen; Ctx, cortex; Cer, cerebellum; Hip, hippocampus; Hth, hypothalamus; LC, locus ceruleus; NA, nucleus accumbens; OTu, olfactory tubercle; SN, substantia nigra; Tha, Thalamus; VTA, ventral tegmental area.)
Regulation of RGS Protein Expression by Abused Drugs
As RGS proteins negatively regulate GPCR signaling, dynamic control of RGS protein transcription would provide a method of controlling the duration and/or intensity of GPCR signaling. It is thus conceivable that drug-induced changes in RGS protein levels over a long time scale could contribute to the chronic effects of tolerance, dependence, and sensitization, and changes in expression on a very short time scale could alter acute responses. Indeed, there is evidence that rapid change in certain RGS protein levels can occur. Selective upregulation of RGS2 mRNA, for example, occurs in the rodent cortex, hippocampus, amygdala, and striatum following exposure to stimuli that induce synaptic plasticity, such as electroconvulsive shock (34), suggesting that RGS2 may thus be modulated as an intermediate early gene product.
The mRNA for RGS2 is also rapidly and selectively upregulated by the psychostimulant drugs amphetamine, methamphetamine, and cocaine (34, 68, 69); again, this upregulation occurs on the immediate early time frame that characterizes the induction of c-fos by these drugs. RGS3 mRNA is also upregulated by stimulant treatment, but at a longer time scale, suggesting a role for the RGS3 protein in adaptation to more sustained stimulation (68). Furthermore, the induction of RGS2 and RGS3 persists with repeated amphetamine administration, possibly reflecting a role in the neuroadaptive changes involved in dependence and sensitization (70).
The effectiveness of amphetamine in inducing RGS2 expression is perhaps due in part to a higher level of dopamine in the striatum following amphetamine administration; a role for dopamine in the upregulation of RGS2 is in fact corroborated through the use of dopaminergic ligands. Amphetamine-induced expression of RGS2 is blocked, for example, by the D1 antagonist SCH 23390 (69). In contrast, the D2 antagonists raclopride and haloperidol stimulate RGS2 mRNA expression and show an additive effect with amphetamine, whereas the D2 agonist quinpirole inhibits RGS2 expression (34, 68, 69). These dopaminergic and drug-induced patterns of RGS2 expression are very similar to patterns of c-fos expression (71).
Some of the molecular mechanisms related to dopaminergic control of RGS protein expression are detailed in Figure 3⇓. The gene for human RGS2 contains a cAMP response element (CRE) (72), and although the promoter for the corresponding rat gene does not contain a CRE, its transcription is still cAMP-dependent (73). Dopamine D1 receptors are positively coupled to adenylyl cyclase; direct stimulation of adenylyl cyclase induces RGS2 expression in several systems (69, 74). Thus, the administration of stimulants increases synaptic dopamine, and postsynaptic D1 receptors thereby stimulate adenylyl cyclase, leading to activation of PKA, CREB phosphorylation, and transcriptional activation of the RGS2-encoding gene. In contrast, the D2 receptor is negatively coupled to adenylyl cyclase, and so agonism of the D2 receptor will ultimately reduce RGS2 expression, particularly because RGS2 mRNA is short-lived (69, 72). Nevertheless, it is an upregulation of RGS2 mRNA that is seen on stimulant administration, and thus a differential distribution of D1 and D2 receptors, on striatal nigral neurons and striatal pallidal neurons, respectively, may explain the additive effects of amphetamine and D2 antagonists (75). On the other hand, because D2 agonists decrease dopamine turnover and dopamine release, and yet the overall effect of amphetamine is to upregulate expression of RGS2 (Figure 3⇓), it is possible that antagonists of presynaptic D2 receptors promote dopamine release and thereby activate D1 receptors. Indeed, concomitant application of D1 and D2 antagonists prevents haloperidol-induced expression of RGS2 (69).
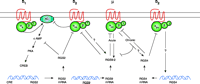
Potential interactions among dopamine or μ-opioid receptors and RGS2, RGS4, and RGS9-2. Because amphetamine and cocaine increase synaptic levels of dopamine in the striatum, dopaminergic receptors are activated indirectly by these drugs. Opioids act directly on the μ receptor. Arrows indicate positive interactions; blunted arows indicate inhibition. The figure is not meant to imply that all receptors are expressed in the same cell.
Although RGS2 is rapidly induced and may thus modulate acute signaling by drugs of abuse, the short half-life of RGS2 mRNA (70) indicates that the protein is unlikely to be a long-term mediator of neuronal changes associated with drug addiction; RGS2 is thus considered to function primarily as a GAP for Gαq (Table 1⇑) (33). Interestingly, several days after binge cocaine treatment of rats, Gαq and Gα11 are upregulated in the paraventricular hypothalamus and amygdala, but not the frontal cortex, and this upregulation persists for several days (76). It is possible that the increase in Gα levels may counteract signaling deficits arising from the upregulation of RGS2 in certain brain regions.
Like RGS2, there is evidence that RGS4 is also highly regulated. In rats, administration of corticosterone (or chronic unpredictable stress) can increase levels of RGS4 in the locus ceruleus and decrease levels in the paraventricular hypothalamus and pituitary (77). Drugs of abuse also induce RGS4 expression, although the effects are not so marked as with RGS2 (Figure 3⇑) (69, 78–80). Curiously, in one study, the level of mRNA for RGS4 in morphine-treated mice did not correlate with changes in RGS4 protein levels. Levels of RGS4 protein were increased twofold by chronic morphine and returned to control levels after precipitated withdrawal, whereas the RGS4 mRNA was unchanged by chronic morphine, but was elevated in the locus ceruleus on withdrawal (80). There is evidence from investigations of corticosteroid-treated mouse AtT20 cells that changes in RGS4 levels may reflect changes in the stability of mRNA for RGS4 combined with a very short half-life of RGS4 protein (81). Under this scenario, the elevated level of RGS4 protein in chronic morphine-treated animals could be explained by stabilization of the protein in the continued presence of drug; on withdrawal from morphine, RGS4 protein would undergo increased degradation, and levels of RGS4 mRNA would coincidentally rise. Consequently, control of degradation may be a significant means of regulating cellular levels of RGS4 protein.
In contrast to the marked upregulation of RGS2 mRNA, a single dose of amphetamine to rats has been reported to cause a downregulation of RGS9 mRNA in striatum (68) or no change (69). However, a small degree of upregulation of RGS9-2 protein was observed in the nucleus accumbens and caudate putamen following chronic administration (and especially chronic self-administration) of cocaine, suggesting a homeostatic response to continual exposure to the stimulant (82). Notably, this increase in protein was specific and was not accompanied by a change in RGS9 mRNA levels (Figure 3⇑); there were no changes in levels of RGS7 or RGS11, which are structurally similar to RGS9 and are considered to be in the same subfamily (R7). In contrast to the stimulants, acute morphine treatment of mice raised the level of RGS9-2 protein approximately 50% within two hours, particularly in the nucleus accumbens, caudate putamen, and regions concerned with the antinociceptive actions of morphine (i.e., dorsal horn of the spinal cord and periaquaductal gray matter). On more chronic exposure to morphine, there was a 50% reduction in the levels of RGS9-2 protein (29). Because changes in RGS9-2 protein do not appear to be regulated by changes in mRNA levels, post-translational effects or regulation of transcriptional rates control the activity of these proteins and the rapidity of their regulation (Figure 3⇑).
RGS Protein Functionality in Response to Drugs of Abuse
Studies devised to define roles for RGS proteins in cell signaling and subsequent animal behavior have used two approaches. The first of these has employed antisense or RNAi techniques and transgenic knockout mice that do not express a given RGS of interest. A disadvantage of this approach can arise if the targeted RGS is not relevant to the physiology being studied, or if non-targeted RGS proteins are compensatory. The second approach employs RGS-insensitive mutants of Gα proteins. Specifically, the functional binding site for RGS4 on the surface of Gαi1 is limited to well-definable regions (83), and only two amino acid residues at the protein surface are essential for high-affinity binding to RGS proteins (84). Mutation of Gly184 of Gα, for example, prevents interaction with RGS proteins and precludes their GAP activity. Such RGS-insensitive Gα mutants can be used to study the physiological consequences of RGS activity without requiring knowledge about which particular RGS protein is involved or identification of RGS function that is independent of GAP activity.
Knockout Mice Deficient in RGS Proteins
The identification of the almost exclusive expression of RGS9-2 in striatal regions associated with the activity of drugs of abuse (Figure 2⇑) led to the development RGS9 knockout mice. Confirming a role for RGS9-2 in response to abused drugs, these mice show much more potent responses to cocaine (82) and morphine (29) than do their wild-type littermates, including behaviors that are associated with abuse liability. In a conditioned place preference assay, mice are trained to associate one side of a chamber with administration of drug (or vehicle); preference for the drug-associated chamber is interpreted as a rewarding effect. Following a moderate dose of cocaine, mice devoid of RGS9-2 protein show an increased degree of place conditioning. The effect with morphine is more marked, such that the null mice show a tenfold preference for the morphine-paired environment (Figure 4A⇓). In this assay, morphine shows a U shaped dose-effect curve with wild-type mice because higher doses do not produce a place preference. In the RGS9 knockout mice, this U-shaped curve is preserved, showing that both aspects of morphine’s action are similarly enhanced. The increased potency of morphine is reversed by restoration of RGS9-2 protein levels in the nucleus accumbens (Figure 4⇓). Interestingly, in morphine-naive wild-type animals, the μ-opioid receptor antagonist naloxone causes a place aversion (i.e., a preference for the non-drug associated chamber), which is more severe in the RGS9 knockouts. The effect of naloxone is possibly due to a blockade of endogenous opioid–tone, which is likely to be higher in the RGS9 knockout mice (Figure 3⇑). To date, experiments with cocaine and morphine in RGS2 knockout mice, similar to those carried out with the RGS9 null mice, have not been reported; however, RGS2 has the potential to regulate both dopamine D1 signaling, via direct inhibition of adenylyl cyclase (89), and D2 receptor signaling by its GAP activity towards Gαi/o (34). Thus, an increase in RGS2 might be predicted to modulate all behaviors associated with stimulant-induced increases in synaptic dopamine levels (Figure 3⇑).
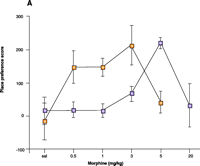
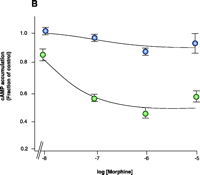
Experimetnal obliteration of the RGS–Gα interaction to study RGS function. A. RGS9 null mice (yellow squares) show increased responsiveness to the rewarding actions of morphine in a place preference assay compared to wild-type littermates (purple squares). (Sal, saline vehicle.) B. Enhanced potency and efficacy of morphine in cells expressing the μ-opioid receptor and either Gao that is insensitive to the action of RGS proteins (green circles) or RGS-sensitive Gαo protein (blue circles). The cells employed in the study (rat glioma C6 cells) do not appear to express RGS9 or other members of the R7 family (i.e., RGS7 and RGS11). Both types of Gαo protein are resistant to pertussus toxin, which is used in all cell cultures in order to inhibit endogenous Gαo activity. Note that the ordinate values for both lines is 1.0.
RGS-Insensitive Mutant Gα Proteins
The increased potency of morphine in the absence of RGS activity has been confirmed in vitro with the use of RGS-insensitive Gαo proteins expressed in rat glioma C6 cells that express the μ-opioid receptor. In these experiments, cells are treated with pertussis toxin (PTX) to uncouple endogenous Gα proteins from the μ-opioid receptor; functional, PTX-insensitive Ga subunits that are either RGS-sensitive (i.e., control) or RGS-insensitive are then expressed in the cultured cells. Cells that express the RGS-insensitive Gαo subunits manifest markedly less adenylyl cyclase activity in response to morphine treatment than do cells expressing Gαo that is sensitive to RGS activity (Figure 4B⇑). In accordance with these results, overexpression of RGS2 shifts the concentration effect curve for morphine to the right for amphibian dermal melanophores that transiently express a μ-opioid receptor (85), and knockdown of several different RGS proteins is reported to modulate morphine antinociception in mice (86). The simplest explanation for these findings is that any RGS-effected GAP activity enhances morphine agonism; however, this simplistic view may not be correct, because the introduction of RGS4 into the nucleus accumbens of RGS9 knockout mice does not alter the place preference response to morphine (29). Moreover, signaling pathways may be differentially affected by RGS action (26). Because the relatively small RGS4 protein comprises mainly the consensus GAP domain, other regions of the RGS9-2 molecule (Table 1⇑) may prove essential to GAP specificity and other protein–protein interactions. Nonetheless, the importance of RGS activity in the negative regulation of opioid signaling is striking, and across neuronal systems, different RGS proteins may serve multiple functions, some of which may be redundant.
Mice deficient in RGS proteins have also been used to investigate sensitization, dependence, and tolerance, all of which are important aspects in the maintenance of drug-seeking behavior. Rodent locomotor activity in response to repeated stimulant administration, for instance, can give a measure of drug sensitization. Relative to wild-type animals, mice devoid of RGS9 show increased locomotor activity in response to amphetamine and cocaine, as well as potentiated locomotor sensitization on repeated cocaine administration. In accordance with these findings, over-expression of RGS9-2 in the nucleus accumbens of rats attenuates the locomotor-stimulating effects of cocaine, although this attenuation is surmountable with higher doses of the stimulant. Unilateral overexpression of RGS9-2 in the nucleus accumbens of rats confirms a role for RGS9-2 in reducing dopaminergic signaling (82).
Administration of increasing doses of morphine to mice induces dependence and withdrawal behaviors on cessation of drug administration or on administration of the antagonist naloxone. RGS9 knockout mice show a significant increase in withdrawal behaviors, such as jumping, wet-dog shakes, paw tremor, diarrhea, ptosis, and tremor. In addition, biochemical markers of withdrawal are increased, including exaggerated upregulation of c-fos in the locus ceruleus. In agreement with this observation, but again providing a discrepancy on the specificity of RGS9-2 for modulating morphine’s effects, C6 cells expressing the μ-opioid receptor and an RGS-insensitive Gαo protein manifest increased adenylyl cyclase “sensitization” to chronic morphine treatment (87). In mice with reduced or obliterated RGS levels (29, 86), anti-nociceptive tolerance to morphine is delayed but not prevented, suggesting a role for RGS proteins in the onset of tolerance. Given the role of RGS proteins as negative regulators of G protein signaling, an early increase in RGS9-2 levels would inhibit acute morphine signaling, and the subsequent decrease in RGS9-2 activity would serve to maintain signaling in the presence of chronic morphine and thus delay tolerance (Figure 3⇑). On the other hand, studies in C6 glioma cells show that endogenous RGS proteins serve to protect against tolerance development to μ-opioid agonists (88). Clearly, observed RGS activity will depend the system studied, the types of RGS proteins expressed, and their selectivity for particular Gα protein-effector combinations.
RGS Proteins as Potential Targets in the Treatment of Drug Abuse
The above discussion has demonstrated roles, or potential roles, for RGS proteins in aspects of the pharmacology of drugs of abuse. Expansion of these studies will help to define roles for specific RGS proteins in cocaine and opioid reward and will also elucidate the functions of specific domains within RGS proteins. The question arises as to whether this information will be useful in developing medications for the treatment of drug abuse.
Information from studies with RGS-insensitive Gα proteins and studies in RGS-knockout mice tell us that by enhancing RGS proteins, we might see a decrease in all behaviors associated with stimulants and opioids. Given the important role for RGS9-2 in attenuating the abuse liability for both morphine and cocaine, it is a potential therapeutic target; it is upregulated by cocaine and amphetamine and could reduce signaling through D1 receptors by a direct inhibition of both adenylyl cyclase and Gαi-linked D2 receptor signaling.
Increasing the activity of RGS proteins is likely to be pharmacologically more difficult than inhibiting their activity. One approach might be provided by agents that selectively limit the degradation of RGS9-2 and RGS2. Interfering with processes that physiologically alter RGS activity, such as phosphorylation (50, 51), protein binding (90), or inositol lipid binding (56–58), could also be used to counteract acute drug abuse. Alternatively, pharmacological agents that act at other receptors might be used to increase levels of these RGS proteins. Indeed, it can be speculated that an increase in RGS protein levels might be a contributor to the potential usefulness of dopamine D1 and D2 ligands as therapeutic agents (91).
An alternative, at least with the opioids, might be to selectively reduce RGS protein activity that inhibits beneficial effects such as antinociception without altering RGS protein functions that limit abuse liability. Such a scenario is supported by evidence that a variety of RGS proteins alter the animal pharmacology of morphine (86) and that the actions of morphine are negatively regulated by RGS proteins other than RGS9-2 in vitro (85, 87, 88). Finally, in RGS9 knockout mice there is a tenfold increase in sensitivity to place conditioning, but only a threefold enhancement of the antinociceptive action of morphine (29). RGS4 is highly expressed in thalamus and brain stem regions (61) that contain high levels of μ-opioid receptors, whereas these regions express none or very little RGS9. RGS4 is also expressed at much higher levels in dorsal root ganglion cells than in other tissues (92), where it could function in the relay of sensory input to the cortex from the periphery. Furthermore, RGS4 is upregulated in the rat lumbar spinal cord in chronic pain states (93), which may contribute to desensitization to morphine in chronic pain, although, paradoxically, RGS4 is downregulated in dorsal root ganglion cells (92). Consequently, an inhibitor that targets RGS4 might increase the therapeutic utility of morphine and related analgesic agents without increasing the rewarding properties that are likely modulated by the striatally restricted RGS9-2. Also, because antinociceptive μ-opioid receptor occupancy could be reduced by broadly inhibiting RGS protein activity, specific targeting of RGS4 would likely limit side effects.
Analysis of the co-crystal structure (83) provides a basis for the rational design of inhibitors of the RGS4–Gαi1 interaction, which should increase the activity of morphine in those cells where RGS4 is the prime negative modulator of morphine signaling. The functional binding site for RGS4 on the surface of Gαi1 is formed by residues in the three switch regions of Gαi1 (i.e., conformationally flexible regions associated with the transition from the GDP-bound to the GTP-bound form). Using this information, a μM-affinity peptide inhibitor of the GAP activity of RGS4 has been synthesized (94). The selectivity of this inhibitor for other Gα–RGS interactions is unknown, but because RGS proteins have only limited homology even within the RGS domain, it may be possible to design selective inhibitors.
Inhibitors that prevent the interaction between Gα and RGS protein have been termed “A-site” inhibitors (22). In contrast, the “B site” represents a second surface within the consensus RGS domain region that binds phosphoinositides and calmodulin. Phosphatidylinositol-3,4,5-triphosphate inhibits RGS GAP activity, whereas Ca2+/calmodulin competes for the InsP3 binding site and stimulates activity (56–58). Consequently, it is theoretically possible to design B-site modulators of GAP activity. Finally, in the more complex RGS proteins, there are functional domains outside of the conserved RGS domain, which carry out scaffolding and/or targeting functions (Table 1⇑) (18) and could provide additional pharmacological targets. High-throughput screens are beginning to identify small-molecule inhibitors of RGS protein function (95).
Conclusions
There is growing evidence that drugs of abuse alter the expression of RGS proteins, and that these proteins modulate the rewarding effects that contribute to continual drug use. In particular, there is compelling evidence of a role for RGS9-2 as a negative regulator of the abuse liability properties of both opioids and stimulants. Consequently, pharmacological agents that target this protein might alter behavioral responses to opioids and stimulants. Nevertheless, cell signaling is highly complex, and there is a myriad of accessory proteins and protein–protein interactions whose precise functions in drug signaling have yet to be elucidated. For example, the mammalian RGS protein family has at least twenty members that can inhibit GPCR signaling. As we learn more about RGS proteins—especially about their various functional domains and their distribution and regulation—and other accessory proteins that modulate GPCR signaling (96), we should be able to identify particular RGS protein targets for medication development.
Acknowledgments
We thank NIDA (DA 04087) and NIH (GM39561) for support, Becky Roof and Jeffrey Talbot for help with the figures, and David Siderovski for helpful comments on Table 1⇑.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Richard Neubig, MD, PhD is Professor of Pharmacology at the University of Michigan Medical School. Rick is one of the pioneers of RGS proteins and in particular their roles as pharmacological targets. When not thinking about RGS proteins he can be found bird watching.

John Traynor, PhD, is Associate Professor of Pharmacology at the University of Michigan Medical School. He has a life-long fascination with the pharmacology and chemistry of opioids. When not fussing with these molecules he can be seen riding his motorcycle. Address correspondence to JRT. E-mail jtraynor{at}umich.edu; fax 734-763-4450.

